Immunology2-
Innate_Humoral_And_Cell_Mediated_Immunity
Immunity to microorganisms is more or less inbuilt which performs at different levels. The human body itself has protection in the form of epithelial layers of cells, skin and otherER-chloroplast pair, those tight associations are involved in bidirectional lipid trafficking between the two compartments. to protect the entry of microorganisms. Attack against invaded pathogens can preventive and immediate within few minutes to hours, which is called innate immunity (inborn/built-in). The innate immunity also triggers cellular structures to act on the viruses or bacteria or parasites, this is called adaptive immunity (consists of Humoral and Cell mediated Immunity).
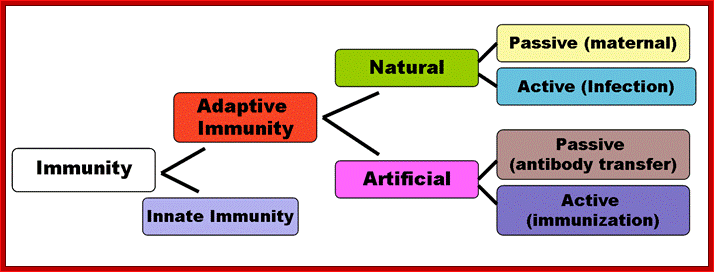
Different forms of immunity; http://en.wikipedia.org/
Innate Immunity:
Innate immune system in-born and it provides the first line of host defense against microbes. The mechanism of innate immunity exists before exposure to microbes. The innate immunity system uses pattern recognition receptors to recognize structures that are shared by microbes that are not present on mammalian cells, and they are often essential for survival of microbes; thus, limiting the capacity of microbes to evade detection. More to it is the epithelial cell layers that cover various organs such as intestine, skin urinogenital organs, ears and eyes etc do have anti-microbial cells and enzymes that take care the invading pathogens immediately aftermath of infections.
The First Lines of Defense;
- Infection->recognize non-specific preformed factor--->Removal of Infectious agents
- Infection->recruitment of effector cells->recognition of PAMPs-> removal of pathogens.
- Infection-> transport of antigens to lymphoids ->Recognition by naïve B and T cells -> Clonal expansion
The response to an initial infection occurs in three phases. These are the innate phases, the early induced innate response, and later the adaptive immune response. The first two phases rely on the recognition of pathogens by germline-encoded receptors of the innate immune system, whereas adaptive immunity uses variable antigen-specific receptors that are produced as a result of gene segment rearrangements. Adaptive
Immunity occurs late, because the rare B cells and T cells specific for the invading pathogen must first undergo clonal expansion before they differentiate into effector cells that migrate to the site of infection and clear the infection. The effector mechanisms that remove the infectious agent are similar or identical in each phase.
The components of innate immune system include epithelial barriers, leukocytes (neutrophils, macrophages, and NK cells), circulating effector proteins (complement ins, collectins, penataxins), and cytokines (e.g., TNF-a, Il-1, chemokines, Il-2, type-I IFNs and IFN-g).
Neutrophils and macrophages are phagocytes that kill ingested microbes by producing Reactive Oxygen Intermediates ROIs, Nitric oxide NO and enzymes in phago-lysosome. Macrophages also produce cytokines that stimulate inflammation and promote tissue remodeling at sites of infection. Phagocyte recognizes and responds to microbial products by several different types of receptors, including Toll-like receptors, mannose receptors and G-protein-coupled receptors.
NK cells are lymphocytes that defend against intracellular microbes by killing infected cells and providing a source of macrophage activating cytokine IFN-g. To combat infection, the phagocytes of the innate immune system facilitate many pattern recognition receptors (PRR) to help recognize pathogen-associated molecular patterns (PAMPs) on the surface of pathogenic microorganisms. Proteins involved in microbial pattern recognition include mannose receptor, complement receptors, DC-SIGN, Toll-like receptors (TLRs), the scavenger receptor, CD14, and Mac-1. PRRs can be divided into three classes: 1. signaling PRRs that activate gene transcriptional mechanisms that lead to cellular activation, 2. endocytic PRRs that function in pathogen binding and phagocytosis, and 3. secreted PRRs that usually function as opsonins or activators of complement.
NK cell recognition of infected cells is regulated by a combination of activating and inhibitory responses. Inhibitory receptors recognize class I MHC molecules, because of which NK cells do not kill normal host cells, but kill cells in which class I MHC expression is produced, such as virus infected cells.
Complementation is activated by microbes and products of complement activation promote phagocytosis and killing of microbes and stimulate inflammation. Other plasma proteins of innate immunity include mannose-binding lectin and C-reactive proteins.
Different cytokines of innate immunity recruit and activate leukocytes e.g. TNF Il and chemokines which enhance the microbicidal activities of phagocytes (IFN-g) and stimulate NK cells and T-cells responses (Il-2). In several infections, excess systemic cytokines production is harmful and even may cause death of the host.
Molecules produced during innate immune response stimulate adaptive immunity and influence the nature of adaptive immune responses. Macrophages activated by microbes and by IFN-g produce co stimulators that enhance T cell activation and IL-12 which stimulates IFN-g production by T cells and the development of IFN-g producing effectors T cells. Complement fragments generated by alternative pathways provide second signals for B cell activation and antibody production.
Innate Immune Response:
Components and function of the innate immune system:
A diversity of components makes up the innate immune system, including:
- Physical barriers (tight junctions in the skin, epithelial and mucous membrane surfaces, mucus itself)
- Epithelial and phagocytic cell enzymes (eg, lysozyme)
- Phagocytes (neutrophils, monocytes, macrophages)
- Inflammation-related serum proteins (eg, complement, C-reactive protein, lectins such as mannose-binding lectins and ficolins)
- Surface and phagocyte granule antimicrobial peptides (defensins, cathelicidin, and many more)
- Cell receptors that sense microorganisms and signal a defensive response (eg, Toll-like receptors)
- Cells that release cytokines and inflammatory mediators (macrophages, mast cells, natural-killer cells).
- Binding of pathogens to cell receptors trigger the process of innate immunity. In fact this can be due to PKR, dsRNA activated protein kinases, RNases and Lymphokines activities.
Gene Expression in Response to Viral Infection:
Animal viruses as well as bacteria and other pathogens infect through cell surface receptors or injury or by endocytosis, the pathogens are released into cell cytoplasm or enter into lymph system, rarely enter into blood. The infected cells respond first by using the ds RNA produced during the replication or transcription of the viral genome. The ds RNAs activate protein kinase (PKR), which phosphorylates protein synthesis chain initiation factor eIF-2a at specific serine 51 residues. The phosphorylated eIF-2α binds to eIF-2β for GDP-GTP exchange, but as the eIF-2α is phosphorylated and the eIF-2β binds to alpha tightly and eIF-2α is rendered nonfunctional. Thus all cellular eIF2-alpha factors get sequestered; in this way protein synthesis of the infected cells is halted.
This blocks the protein synthesis in the cell. The ds RNA can also be used as defense mechanism by producing si or mi RNA (RNA interference mechanism). This can also induce the synthesis of Oligo’s which activate RNases that can cleave RNA molecules. Cells infected with pathogens react to generate antiviral immunity and at the same time they also induce to produce Interferons, which on release activate cells to release more IFNs and activate several specific genes so as cell to be in antiviral state; this first defense mechanism called innate immunity, which leads to adaptive or cellular immunity.
|
Physico-chemical barriers to infections |
||
|
System/Organ |
Active component |
Effector Mechanism |
|
Skin |
Squamous cells; Sweat |
Desquamation; flushing, organic acids |
|
GI tract |
Columnar cells |
Peristalsis, low pH, bile acid, flushing, thiocyanate, gastric acid, bile acids, enzymes, mucus, saliva, lysozymes |
|
Lung |
Tracheal cilia |
Mucocialiary elevator, surfactant |
|
Nasopharynx and eye |
Mucus, saliva, tears |
Flushing, lysozyme, defensins, thiocyanate |
|
Circulation and lymphoid organs |
Phagocytic cells NK cells and K-cell LAK |
Phagocytosis and intracellular killing Direct and antibody dependent cytolysis IL2-activated cytolysis |
|
Serum |
Lactoferrin and Transferrin |
Iron binding |
|
Interferons |
Antiviral proteins |
|
|
TNF-alpha |
antiviral, phagocyte activation |
|
|
Lysozyme |
Peptidoglycan hydrolysis |
|
|
Fibronectin |
Opsonization and phagocytosis |
|
|
Complement |
Opsonization, enhanced phagocytosis, inflammation |
|
|
|
|
|
|
|
|
|
|
|
|
|
“The epithelial surfaces form a physical barrier that is very impermeable to most infectious agents, acting as the first line of defense against invading organisms. Desquamation of skin epithelium also helps remove bacteria and other infectious agents that have adhered to the epithelial surfaces. In the gastrointestinal and respiratory tract, movement due to peristalsis or cilia helps remove infectious agents. Also, mucus traps infectious agents. The gut flora can prevent the colonization of pathogenic bacteria by secreting toxic substances or by competing with pathogenic bacteria for nutrients or attachment to cell surfaces. The flushing action of tears and saliva helps prevent infection of the eyes and mouth.”
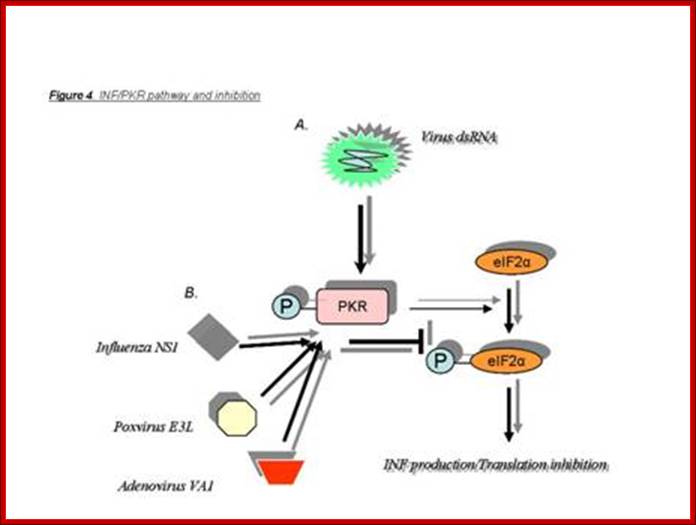
Virus infection leads to IFN and ds mediated PKR reactions as the first defense against viral attacks. http://homepage.usask.ca/
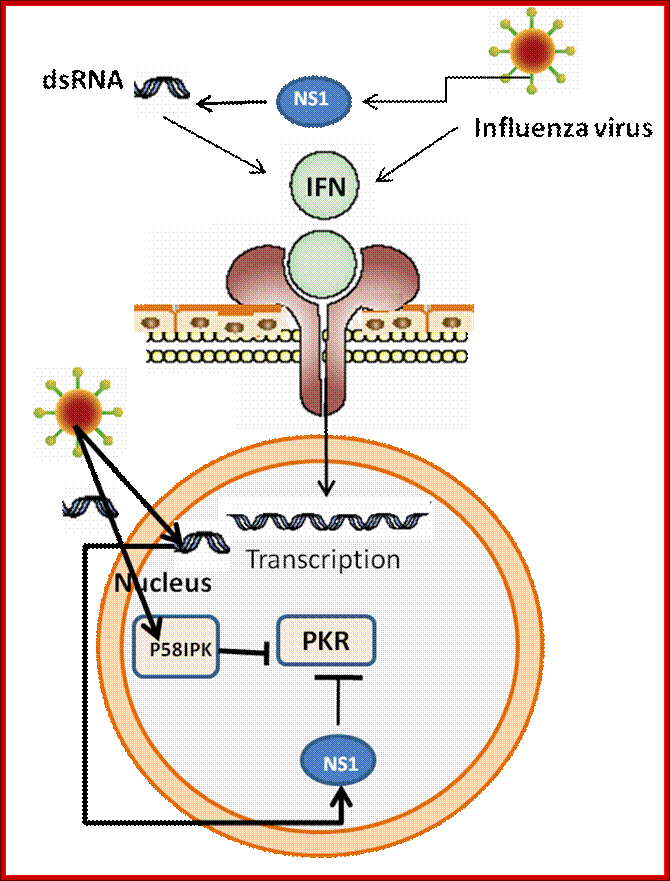
Interferons (α and β). Interferons are cytokines that are produced in small amounts by cells infected with virus. Are efficiently induced by the presence of double-stranded viral RNA (viral replication intermediary), the process involves three antiviral proteins: protein kinase (PKR), Oas1 and RNase L, which block the translation and degradation of viral and cellular RNAs. Interestingly, influenza virus induces high levels of interferon with protective properties; Influenza virus mechanisms to evade interferon action. The, NS1 protein encoded by the virus genome suppresses induction of IFNs-α/β. P58IPK is a cellular inhibitor of PKR that is activated by influenza – virus infection. Modified from: Katze 2002.; http://www.intechopen.com/
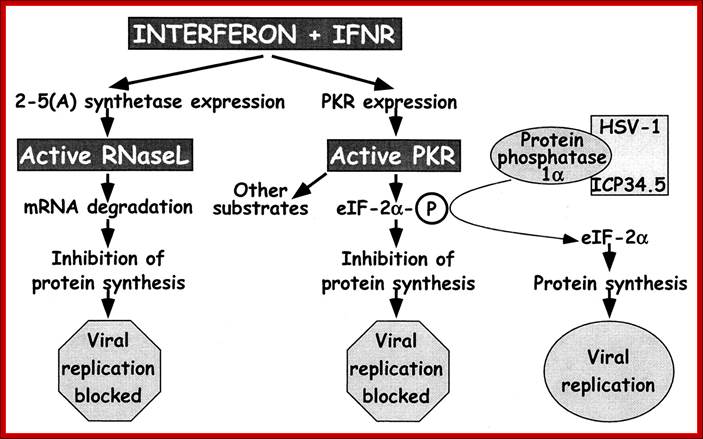
IFN-mediated induction of the antiviral state through independent pathways by RNase L and PKR: For RNase L, IFNs interacting with their receptors lead to expression of 2-5A synthetase, which, after double-stranded RNA binding, produces 2′,5′-oligoadenylates that lead to RNase L activation and blockage of viral replication. IFN also induces expression of PKR, which is also activated through double-stranded RNA binding. One of the activities of PKR is to phosphorylate eIF-2α leading to cessation of protein synthesis and blockage of viral replication. To evade this antiviral mechanism, HSV-1 ICP34.5 binds to protein phosphatase 1α and dephosphorylates eIF-2α, lifting the block on viral replication. Mice with null mutations in the IFN receptors, RNase L, and PKR (gray boxes) were used in this study to dissect the interaction of ICP34.5 with these pathways. http://www.pnas.org/
Immediately after infection, cells respond by innate immunity and make neighboring cells prepared for defense against the viral or any such pathogen attack. In the process the first cell lines infected with pathogens die; self scarifies for the other sister cells.
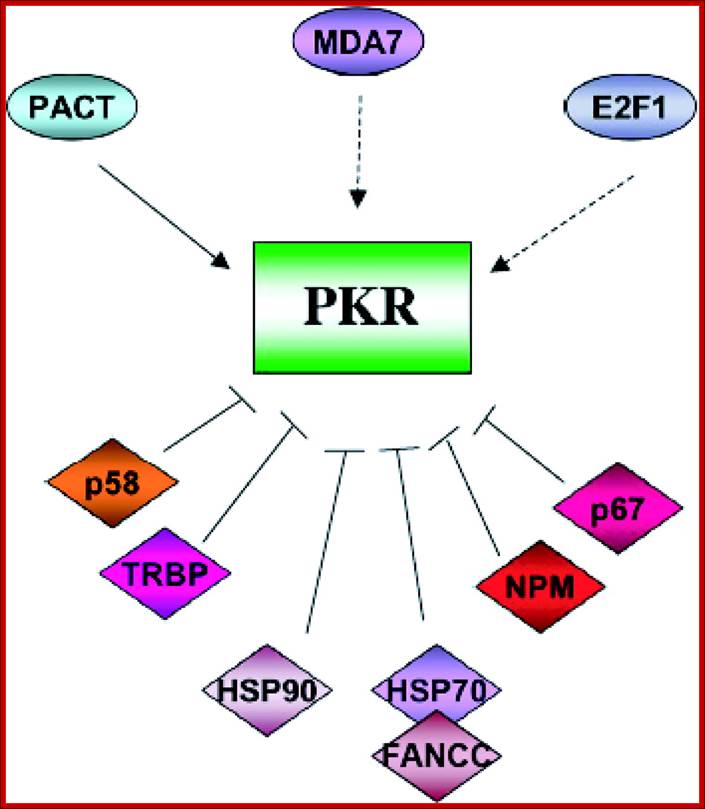
Impact of Protein Kinase PKR in Cell Biology: from Antiviral to Antiproliferative Action; PKR is modulated by a number of cellular proteins; Microbiol. Mol. Biol. Rev. http://mmbr.asm.org/
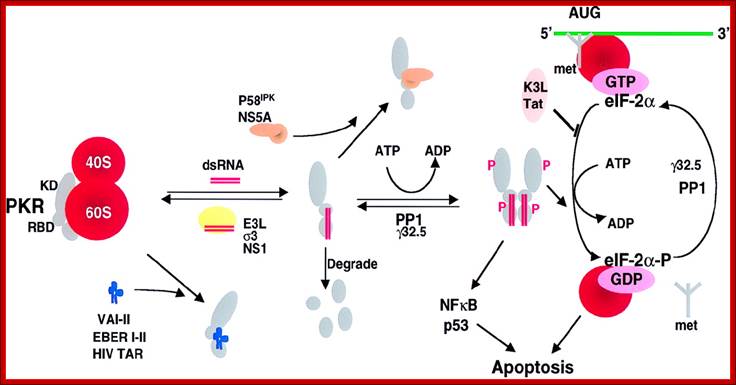
Viral inhibitors of the PKR pathway. PKR is depicted as a monomer with a kinase domain (KD) and two dsRNA binding domains (RBD) bound to the 60S ribosomal subunit. The binding of dsRNA induces a conformational change to promote PKR dimerization, auto phosphorylation, and activation of the eIF-2α kinase activity. Activated PKR also leads to activation of NF-κB, p53, and IFN regulatory factor 1 (IRF1). Viral inhibitors that act through different mechanisms are depicted. Adenovirus VA RNAs, Epstein–Barr virus EBER RNAs and HIV transactivator responsive region TAR RNA bind and inhibit PKR and presumably displace PKR from the ribosome. Numerous viral proteins, such as Vaccinia virus E3L, influenza virus NS1, and Reovirus σ3 bind and sequester dsRNA, thereby preventing activation by dsRNA. Vaccinia virus K3L and HIV trans-activating transcriptional activator Tat act to inhibit PKR binding to eIF-2α. Protein phosphatase PP1 dephosphorylates phosphorylated eIF-2α as well as phosphorylated PKR. Herpes simplex virus 1 encodes a protein γ32.5 that facilitates activation of PP1. Hepatitis C virus nonstructural protein NS5A prevents PKR dimerization. In addition, influenza virus activates a cellular inhibitor P58IPK that also inhibits PKR dimerization. Poliovirus induces PKR degradation. (See ref. 5 and references therein). met represents initiator methionyl tRNA, and AUG represents the initiator codon; http://www.pnas.org/
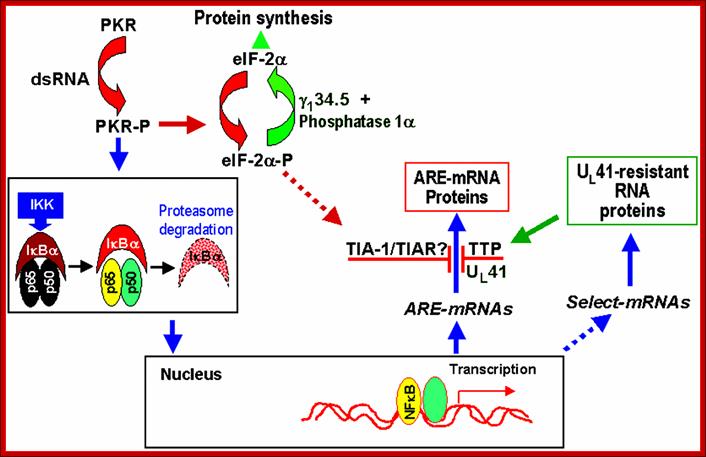
Schematic representation of NF-kB-dependent RNA synthesis and vhs-dependent selective degradation in HSV-1-infected cells; http://www-06.all-portland.net
Double-stranded RNA synthesis during HSV-1 infection leads to the phosphorylation of PKR. The activation of PKR leads, then, to the activation of IKK, which allows IκBα degradation by the proteosomal pathway. The degradation of IκBα releases NF-κB, which can then migrate to the nucleus. NF-κB acts there as a transcriptional activator of numerous genes. Among them, several ARE-containing mRNAs, such as IEX-1, IκBα or c-fos, were found to be up-regulated by HSV-1. The translation of these mRNAs is blocked in HSV-1-infected cells in a UL41-dependent manner. At the same time, selected mRNAs, resistant to UL41 action, are translated during HSV-1 infection. Among these proteins, TTP, which is not activated by NF-κB, is largely expressed after HSV-1 infection and considerably less induced with ΔUL41 deletion mutant. TTP, similar to TIA-1 and TIAR, is an ARE-binding protein involved in the normal degradation pathway of ARE-containing RNAs. TIA-1 and TIAR accumulate in the cytoplasm after HSV-1 infection. / http://www-06.all-portland.net/
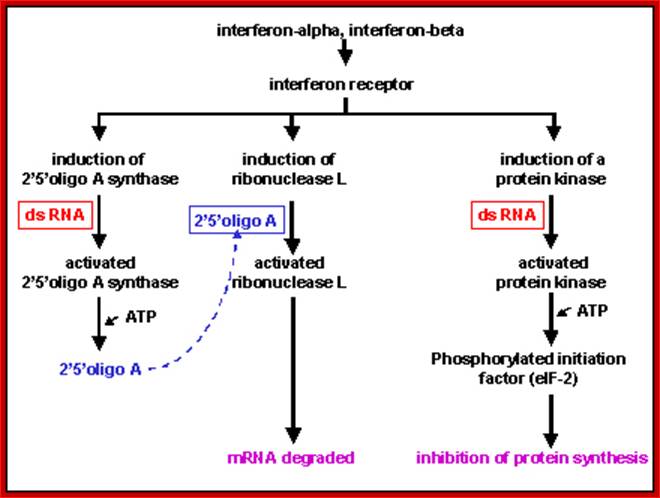
Effects of Interferons INTERFERON-alpha AND INTERFERON-beta (TYPE I INTERFERONS); these interferons induce about 20-30 proteins, and the function of many of these is not fully understood. However, three of the proteins that appear to play an important role in the induction of the anti-viral state have been intensively studied. Expression of one of these proteins (2’5’ oligo A synthase) results in activation of the second of these proteins (a ribonuclease) which can break down mRNA, and expression of the third protein (a protein kinase) results in inhibition of the initiation step of protein synthesis. These activities target viral protein synthesis, but also result in inhibition of host protein synthesis. Thus it is important that these proteins are only made and activated when needed. http://pathmicro.med.sc.edu/
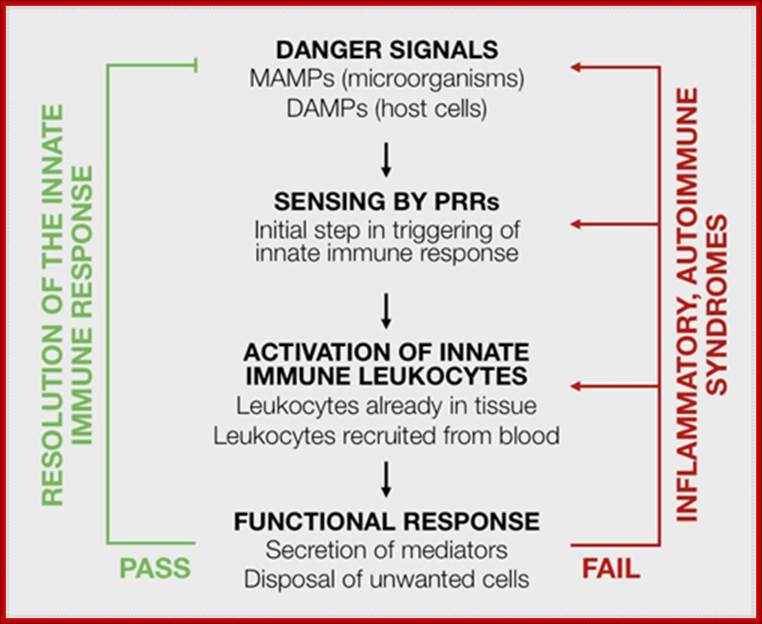
Figure caption: Schematic view of innate immune responses. Danger signals from microorganisms (microorganism-associated molecular patterns, or MAMPs) and host cells (damage-associated molecular patterns, or DAMPs) are key to initiation of innate immune responses, via the activation of pattern recognition receptors (PRRs), which are shared by all members in a species (“innate” receptors). Innate immune leukocytes are then activated and mount a functional response. The ideal outcome (labeled as “pass”, in green) will remove the initial danger signal and be limited in time, space and resources, leading to a resolution of the innate immune response. However, a vicious circle may ensue, with further emission of danger signals, sensing by PRRs and activation of innate immune leukocytes (labeled as “fail”, in red), leading to chronic inflammatory or autoimmune disease. http://www.keywordsking.com
Infection can also lead to inflammation; it is the first response, due to the action of dendrite and macrophage cells and mast cells. These cells contain Pattern Recognition Receptors (PRRs) on their surface (different from host cells), which are collectively called PAMPs. Chemical factors released during inflammation histamines, serotonins, leukotrines and prostaglandins. Neutrophils trigger activities of other leukocytes and lymphocytes. Macrophages activated also release cytokines-such as TNFs, HMGB1 and IL-1.
Interferons:
Historically these were first observed by two Japanese workers in the University of Japan more than (+/-) 57 years ago; they are Yasu-ichi Nagano and Yasuhiko Kojimma. They showed that untreated viral infected skin cells were able to inhibit the second time viral injection. This discovery was published in French ‘Journal dela Societe de Biologie’ (1954). In London Alick Isaacs and Jean Lindermann worked on heat killed flu viruses (1957). When they found resistance effect they called them as INTERFERONS, means they interfere with viral function.
Interferons are glycoproteins produced in response to pathogen infections. The kind of IFNs depends upon the cell types that produce IFNs. There are three kinds of Interferons:
IFN-alpha: Produced by leukocytes, (165a.a) prevent viral DNA or RNA replication, induce MHC1 expression. The gene is located on hu-9p22 chromosome
IFN-beta: It is rapidly produced by fibroblasts, (166a.a), act on macrophages and activate natural killer cells (NKs). Induce MHC I expression.
IFN-gamma: Produced by lymphocytes, (146 a.a), they act on macrophages, activate NK cells. Activate MHCI and MHCII class gene expression. They also activate CD4T helper cells (cluster domain) and CD8T-cytotoxic cells.
Interferons are also grouped into several types such as Type I, Type II and Type III. This classification is based on the structure and functions. They are very effective at low concentration of 10x^14M. They bind to wider variety of targets.
Type I: Consists of Interferon alpha (there are 13 of them) and Interferon beta. Most of them are produced in Leucocytes and fibroblasts and such somatic cell types. They are effectors of innate and adaptive immunity. They activate NK cells, macrophages, MHC class-I antigens, protection to CD8 cells from death
Type II: Consists of Interferon gamma, which is derived from lymphocytes.
Type III: Consists of interferon lambda and few others.
Animal cells have specific receptors for each of the mentioned glycoprotein types. When IFNs bind to receptors, cells respond and develop into antiviral state. Activation of certain genes does this and their products act as defense systems against further viral attack. This acquisition self defense is often called as innate immunity, which leads to Adaptive immunity.
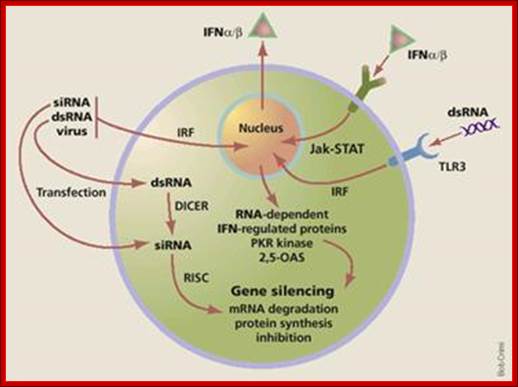
V"C:\Users\grkra\Desktop\Doc 3.docx"
http:www.homepage.usask.ca/;
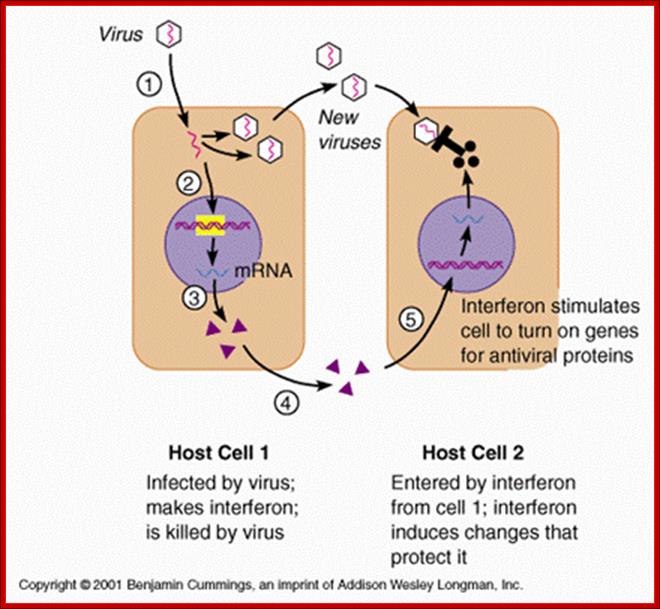
C V"C:\Users\grkra\Desktop\Doc 3.docx"
http:www.homepage.usask.ca/;
A general view of how antiviral state is generated by different factors.
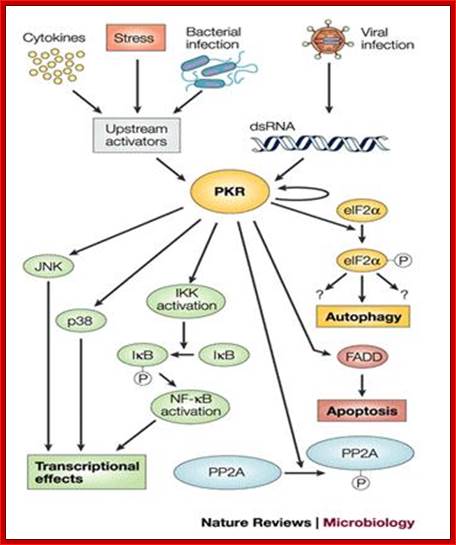
http:www.homepage.usask.ca/;http://www.fotosimagenes.org/
A general view of how antiviral state is generated by different factors.
Protein Kinase PKR has many functions such as Phosphorylating components such as eIF2A, JNK, p38, IKK-IkB, NFkB and PP2A.
Viruses, bacteria and stress factors activate protein kinase which in turn inhibit protein synthesis by phosphorylating eIF2 α and activate apoptosis, JNK, p30, NFkB and PP2A.
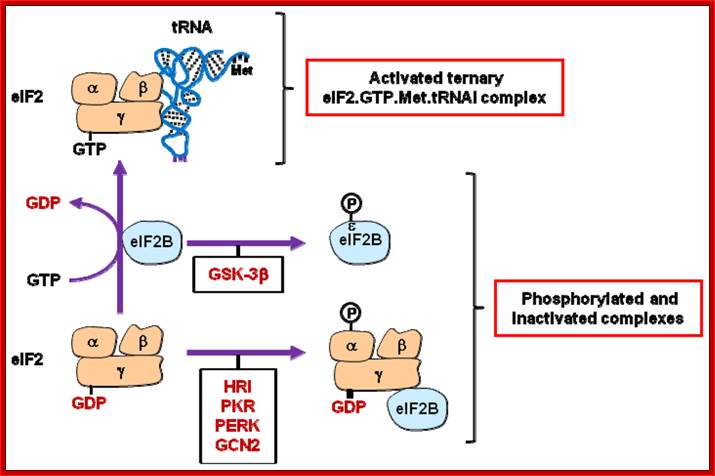
Regulation of eIF2 activity. The guanine nucleotide-exchange activity of eIF2B permits the association of the eIF2γ subunit of eIF2 with GTP. This is a prerequisite for the formation of the ternary eIF2.GTP.Met.tRNAi complex. eIF2 and eIF2B activities are inhibited by phosphorylation of their eIF2α (by HRI, PKR, PERK or GCN2) and eIF2Bɛ (by GSK3β) subunits. eIF2B activity is also indirectly inhibited (sequestered by eIF2γ) as a consequence of eIF2α phosphorylation. See text for details. http://www.mdpi.com/
At the same time some dsRNA sequences can induces the synthesis of 2’-5’Adenine oligo’s. These oligo’s activate RNase L, which simply degrades all RNAs; thus viruses or for that matter any of the other pathogens cannot multiply or propagate. The dsRNA sequences, some, can attract protein complexes such as DiCer; they in turn generate smaller si or mi RNAs which in association with RNA induced silencer complement RISc binds to viral RNAs or mRNAs in sequence specific manner and block translation or degrade the same, which is considered as the most potential RNAi mediated destructive phenomenon. RISc is associated with a group of proteins of which one of them is an RNase.
Viral infection leads to the synthesis of some RNAs containing complementary sequences that fold to generate dsRNA during its genome replication (RNA genome) or its transcripts which in turn activate PKR. Protein kinase C and other protein kinases activated by cell receptors for Lipopolysacharides, TNF (Tumor Necrosis Factors) and T and B cell antigens act on IkB and phosphorylate the same that releases NFkB from the IkB. The active NFkB enters the nucleus and activates several genes as shown in the figure.
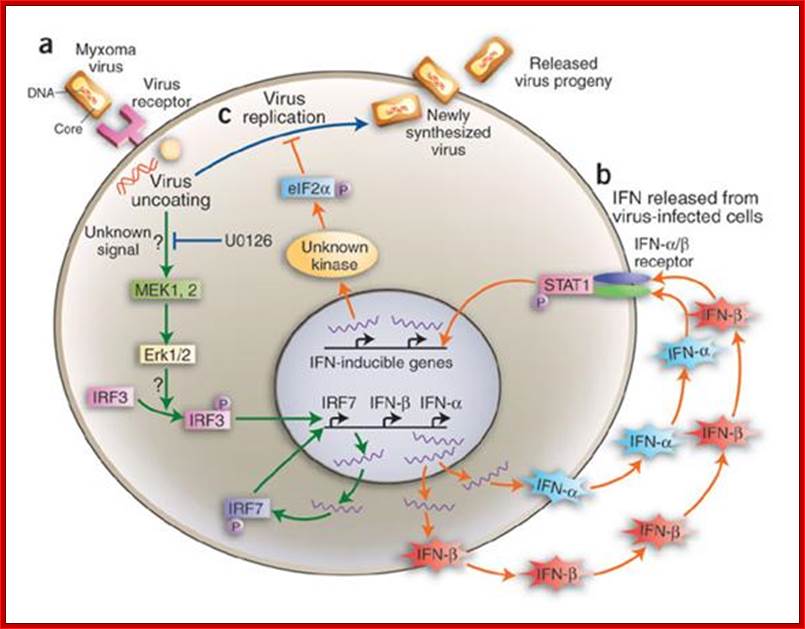
Infection leads to activation of kinases such as MEK, ERK which phosphorylate IRFs (Interferon Regulatory factors). Activated IRFs enters into the nucleus and binds to specific IFN genes and activateIRF7, IFN-b, IFN-a. This leads to the synthesis of more IFNs which on release bind to specific receptors and the receptors activate specific STATs by phosphorylation which activates specific genes. http://www.nature.com/
At the same time viral infection via signal transduction also activates cytoplasmic interferon gene regulatory factors (IRFs) in the cell. The phosphorylated regulatory factors (IRFs) bind to their specific IREs Response elements of specific genes and recruit cofactors such as p300/CBP and activate IFN genes. The interferons synthesized are released into external space. The released IFNs bind to the receptors of neighboring cells and activate cells into antiviral state; this chain reactions result in a cascading effect.
RNA interference:
RNA interference (RNAi) or Post-Transcriptional Gene Silencing (PTGS) is a conserved biological response to double-stranded RNA that mediates resistance to both endogenous parasitic and exogenous pathogenic nucleic acids, and regulates the expression of protein-coding genes. This natural mechanism for sequence-specific gene silencing promises to revolutionize experimental biology and may have important practical applications in functional genomics, therapeutic intervention, agriculture and other areas.
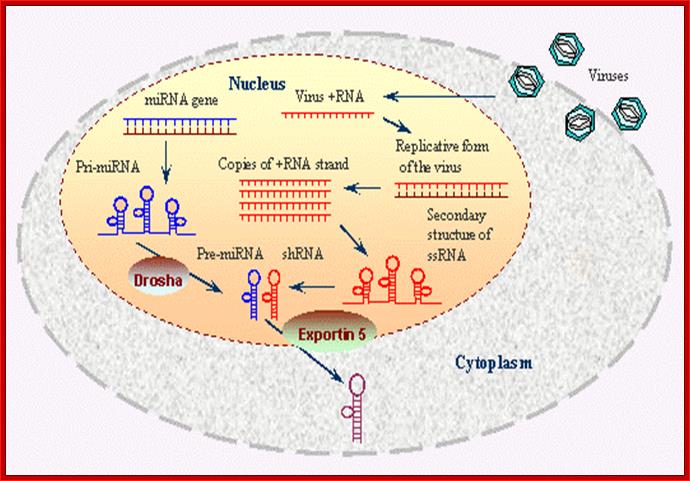
Viral generated ds RNA is used by Dicer and then by RISC protein complexes bind and generate RNAi defense against invading viruses or any other pathogen; http://www.ncbi.nlm.nih.gov/
IFN genes contain certain regulator elements such as VREs. For example activation IFN beta requires a variety of factors such as Nuclear Factor Kappa Beta, NFkB, Jun/ATF and High Mobility Group proteins (HMG). The most important factor is IFN response element binding factor called IRF. There are several IRFs (at least 9 of them) of which IRF3 is important in activating IFN-beta gene. Viral infection induces dsRNA mediated activation of proteins that leads to phosphorylation of IRF3 and IKB (inhibitor of NF kB) the latter is bound to NFkB (dimer proteins). Phosphorylation of IkB, makes the IkB to be released from NFkB and it is destroyed by ubiquitinated process. Then the dimers of NFkB factors move from cytoplasm in to the nucleus. IRF3, NFkB, jun/ATF and HMG proteins bind to their specific sites. High mobility proteins are a family of proteins; bind to the minor groove of specific DNA sequences and induce DNA bending. The binding of these factors recruit cofactors such as CBP/P300 and mediator complexes. The DNA then loops and generates a structure referred to as enhanceosome and activates the IFN gene. The mechanism for other IFN genes is more or less the same except for different IRFs and other cellular factors. IRF3 is involved in activating IFN alpha and beta and IRF and IRF7 is produced in lymphoid cells. Each of these genes have what is called positive regulatory domain (PRD) and negative regulatory domains (NRD). They bound by NRDs before viral infection. Factors bound to NRD domains are released after viral activation and viral PRDs bind and activate the genes. Trans-retinoic acid and IFN-alpha inhibit cell proliferation and induce squamos carcinoma cell apoptosis.
IFN-beta Gene regulatory elements and the factors bound to them:
-HMG-Jun/ATF-(GAAAG/C)3 –CAAT-NFkB-HG—TATA--+1>
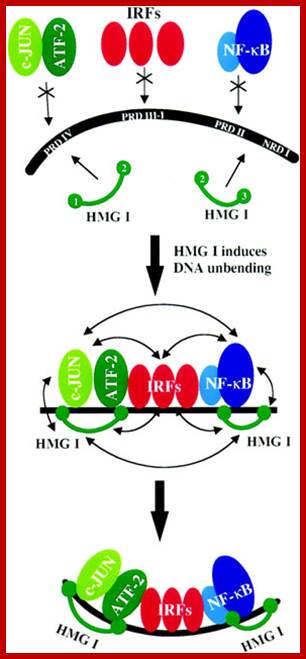
Jun, HMG1, ATF IRFs and NF-kB bind to promoter elements and activate IFNbeta genes. IRF-1 is actually a weak activator and this has to do with the distinct activities of each end of the protein. The N-terminus is important in IRF-1’s binding to DNA whereas the C terminus is important for its activation (2). A single point mutation in codon 8 transforms a methionine to leucine and has been implicated in gastric cancer probably because IRF-1 is unable to bind DNA correctly. Interestingly the protein’s N-terminus has also been found to have a repressive activity with respect to the rest of the protein. In a report by Kirchhoff et al, the researchers found that deleting residues 1-60 of the N-terminus, increased the activity of the protein 240-fold
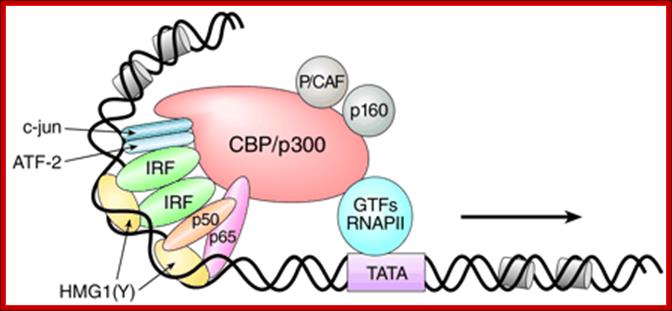
The IFN-{beta} enhanceosome complex. Assembly of the IFN-{beta} enhanceosome creates a stereospecific interaction surface for recruitment of CBP/p300 and the basal transcription machinery to allow multiple rounds of transcription. GTFs indicate general transcription factors. p160 refers to the SRC/TIF/pCIP family of coactivators. http://www.biomed-search.com/
This innate immunity leads to adaptive immunity where T and B- lymphocytes are activated to produce antibodies against pathogen’s antigens.
IFNs perse activate several cellular genes; can be 70-90 different genes. Thus they produce far reaching effects. Each of the IFN regulated genes have elaborates regulatory elements at its 5’ upstream.
The IFNs are also called cytokines, some of them are glycoproteins. They will be circulating in blood stream and activate cells through their specific receptors and render them anti viral.
Interferons are called by that name, because they interfere with the viral multiplication and act as first and foremost defense systems against first and second viral attack. They are produced in response to stress or pathogen. The response to infection is within 2-3hrs. The IFNs released act on the receptors found on several kinds of cells and make them immune to viral attack; this type of response to viruses is called innate response. They are not viral species specific, but they are specific to tissue types; ex. murine tissue is specific to murine cells.
One of the products that is very effective in defense system is double stranded RNA which actually triggers the defense process. Another is dsRNA dependent protein kinase. The third one is 2’-5’oligo synthesis and it activates RNase L. The antiviral state has the following components.
Interferons do activate several cellular genes which have far reaching effects on cells. Cells respond to IFNs through specific receptors at cell surface. In order to activate IFN stimulated genes (ISGs) they require interferon regulatory factors (IRFs).
Interferons released from the infected cells circulate in the extra cellular fluid and act on variety of other cells. Different types of IFNs act on different types of cells, which is determined by the kind of IFN receptors they contain. The binding of IFNs to cell surface receptors is more or less specific. The binding leads to activation of receptor mediated kinase that leads to signal cascade in activating specific genes.
Cytokines are chemical messengers or hormones; they are extremely potent and act at very low concentrations. They are very specific, and act through specific receptors of the target cell membrane.
The cytokine receptors are membrane glycoproteins consisting on several units. They have the role of internalization of the signal. This initiates a cascade of intracellular signaling with many proteins involved. Among them, kinases of the JAK family, which attracts and activate the transcription factors of the STAT family. The result of this chain reaction is the induction of several genes that mediate the biological activities of cytokines. Besides membrane receptors, soluble receptors for different cytokines have been found in sera; they are similar to the membrane receptors and are found in large quantities. It is thought that their function is the regulation of the cytokine production, and to act as antagonists of the membrane receptors.
Type-I IFN-alpha and beta bind to IFNAR receptors and Type-II IFN-gamma bind to IFNGR receptors. Type III-IFN lambda bind to different types of receptors.
Investigations on IFNs receptors have revealed that there are a family of receptor cum protein kinases. Inside one finds another family of transcriptional factors called STATS, meaning Signal Transducers and Activators of Transcription. Binding of IFNs to their cell specific receptors activate receptor endowed with kinase domains; they may be tyrosine or serine or threonine kinases. They in turn activate STATs by phosphorylating specific STATs for there is a family of STATs.
The receptors for type-I IFNs are –IFNAR1/JYK2-IFNAR2/JAK1; they phosphorylate STAT1-STAT2 which interact with IRF9. Receptors for type-II IFNs are IFNGR2/IFNGR1-JAK1-IFNGR1/IFNGR2-JAK1 ; they interact with STA1 and STAT2. Type III IFN bind to receptors lkeIL10R2/Tyk2-IFNLR!/JAK1.
They form a kind of link between cell surface receptors and several gene expressions, ex. IFN beta.
IFN alpha is expressed within 20 minutes of binding to cell surface receptor, induces the expression of some subset of genes by 20 fold.
The genes respond to IFN-alpha contain Interferon Stimulated Response Elements (ISRE) in their regulatory regions.
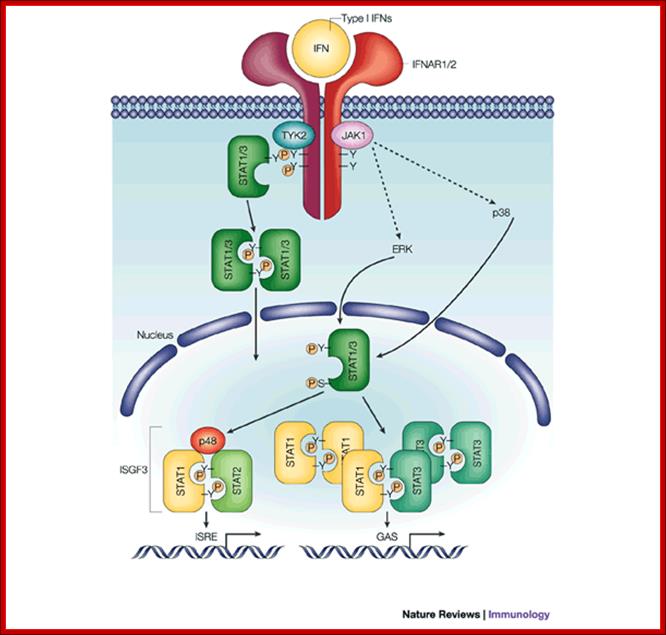
The IFN
receptors and JAK–STAT signalling; Michael G. Katze,
Yupeng He & Michael Gale, JrThe primary
players in the interferon (IFN) signaling pathways are the signal transducers
and activators of transcription (STATs) and Janus kinases (JAKs)129, 130(see figure). STATs are latent
transcription factors that become tyrosine phosphorylated by the JAKs. There are many members of the JAK–STAT family of
proteins that have a role in various combinations in regulating the synthesis
of both IFN /
/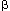 - and IFN-
- and IFN-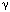 -induced genes131. Type I IFNs bind to their
cell-surface specific receptors (IFNAR1/2) and activate the intracellular IFN signaling
pathway, which involves mainly JAK1 and TYK2,
and STAT1, STAT2 and STAT3.
The JAKs phosphorylate and activate the STATs, which homo- or heterodimerize
and translocate to the nucleus to induce the expression of the IFN-stimulated
genes (ISGs). The STAT1–STAT2 heterodimer complexes with nuclear protein p48,
and this complex binds to the IFN-stimulated response element (ISRE) sequences
in the promoters of ISGs. STAT1 and STAT3 homo- and heterodimers bind to -IFN-activated sequence (GAS)
elements. In addition, the mitogen-activated protein kinase (MAPK) pathways
(involving extracellular-signal-regulated kinase (ERK) and p38 MAPK) have a
role in IFN signaling, by phosphorylating serine residues of STAT1 and STAT3
and further enhancing their transcriptional activity. Type II IFN signaling
follows a similar, but distinct, pattern. The events that are responsible for
driving transcription from the ISRE — the cis-acting DNA element that is found in IFN-/-inducible genes — are important for
virus regulation. For IFN-/, phosphorylated STAT1–STAT2
heterodimers are recruited to ISRE sequences together with IFN-regulatory
factor 9 (IRF9) or p48 to form ISGF3, which is a multi-component transcription
complex. This regulation is distinct from that of the IFN- pathway, which relies on STAT1 homodimers for
activation through the GAS element. Most of the components of these complex
signalling pathways have been targeted for disruption in experiments using
knockout mice. These mouse experiments have validated the importance of these
host pathways in fighting virus infection. http://www.nature.com/
-induced genes131. Type I IFNs bind to their
cell-surface specific receptors (IFNAR1/2) and activate the intracellular IFN signaling
pathway, which involves mainly JAK1 and TYK2,
and STAT1, STAT2 and STAT3.
The JAKs phosphorylate and activate the STATs, which homo- or heterodimerize
and translocate to the nucleus to induce the expression of the IFN-stimulated
genes (ISGs). The STAT1–STAT2 heterodimer complexes with nuclear protein p48,
and this complex binds to the IFN-stimulated response element (ISRE) sequences
in the promoters of ISGs. STAT1 and STAT3 homo- and heterodimers bind to -IFN-activated sequence (GAS)
elements. In addition, the mitogen-activated protein kinase (MAPK) pathways
(involving extracellular-signal-regulated kinase (ERK) and p38 MAPK) have a
role in IFN signaling, by phosphorylating serine residues of STAT1 and STAT3
and further enhancing their transcriptional activity. Type II IFN signaling
follows a similar, but distinct, pattern. The events that are responsible for
driving transcription from the ISRE — the cis-acting DNA element that is found in IFN-/-inducible genes — are important for
virus regulation. For IFN-/, phosphorylated STAT1–STAT2
heterodimers are recruited to ISRE sequences together with IFN-regulatory
factor 9 (IRF9) or p48 to form ISGF3, which is a multi-component transcription
complex. This regulation is distinct from that of the IFN- pathway, which relies on STAT1 homodimers for
activation through the GAS element. Most of the components of these complex
signalling pathways have been targeted for disruption in experiments using
knockout mice. These mouse experiments have validated the importance of these
host pathways in fighting virus infection. http://www.nature.com/
Four proteins have been found to bind to ISRE and their genes have been cloned.
STAT-alpha is 91 KD protein, STAT-beta is 84kd protein, and they are identical with exception that Stat1-alpha is 30 amino acids longer at C-terminal. The same gene codes both, but by alternate splicing they generate two different polypeptides.
The third is STAT-2 is 113kd protein. This protein shows 59% homology with stat 1-alpha and stat 1-beta.
All these proteins have SH2 domains for protein-protein interaction to dimerize to produce homodimers or heterodimers among them.
The fourth protein, called P48 is actually the DNA binding protein and it is unrelated to the first three stats and it has no SH2 domains.
Only those Cells, which are stimulated by IFN-alpha, contain the above-mentioned three STATS, which are found in cytoplasm.
On the contrary, P48, whether the cell is stimulated or not, it is found both in cytosol and nucleus. The P48 can bind to DNA to specific ISRE elements even without signals and the binding is independent of STATS.
The receptors for IFNs, all the three, belong to a super family of receptors.
They don’t have intrinsic protein kinase activity, but the cytosolic domain of the receptor on binding to the ligand gets activated. In this state they associate with one or more cytosolic protein kinases and activate them. But receptors without ligand binding don’t do so.
The activated IFN receptor is believed act as TYK-2 and JAk-1 (tyrosine kinase-2 and Janus kinase-1).
The activated TYK-2 or JAK-1 phosphorylates STATS at Tyrosine residues.
Phosphorylated STATS interact with one another and generate heterodimers (one with phosphorylated protein dimerizes with another containing SH2 group). The dimerized STATs move in to the nucleus and interact with P48, which is already bound to ISRE; this leads to activation of the transcriptional apparatus.
Each type of IFNs induces unique subset of genes by using IFNs-STATS signaling pathway. This specificity stems from the fact that each receptor is different and their associated proteins are different and the STATS are different and their ISRE elements may be different but may be located in different positions.
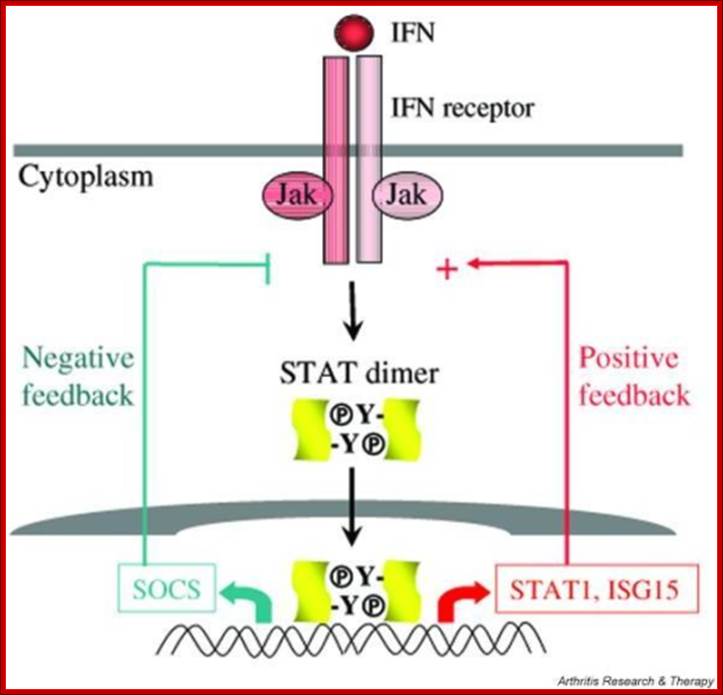
A variety of cytokines and growth factors use the Janus kinase (Jak)-STAT signaling pathway to transmit extracellular signals to the nucleus. STATs (signal transducers and activators of transcription) are latent cytoplasmic transcription factors. There are seven mammalian STATs and they have critical, nonredundant roles in mediating cellular transcriptional responses to cytokines. The physiological roles of STATs have been elucidated by analysis of mice rendered deficient in STAT genes. STAT activation is regulated and can be modulated in a positive or negative fashion; it can be reprogrammed to drive different cellular responses. Several auto-regulatory and signaling crosstalk mechanisms for regulating Jak-STAT signaling have been described. Understanding and manipulation of the function of STATs will help in the development of therapeutic strategies for diseases that are regulated by cytokines. http://openi.nlm.nih.gov/
Specificity stems from cell lines, e.g. Mutants for TYK-2 are insensitive to IFN-alpha, but are sensitive to IFN-gamma, which suggests that the TYK-2 is a component of IFN-alpha specific pathway.
Cell line mutants for JAK-1 are sensitive to both IFN-alpha and IFN-gamma, thus they play a role in both pathways. However IFN-gamma receptors have shown to be associated with second kinase called JAK-2.
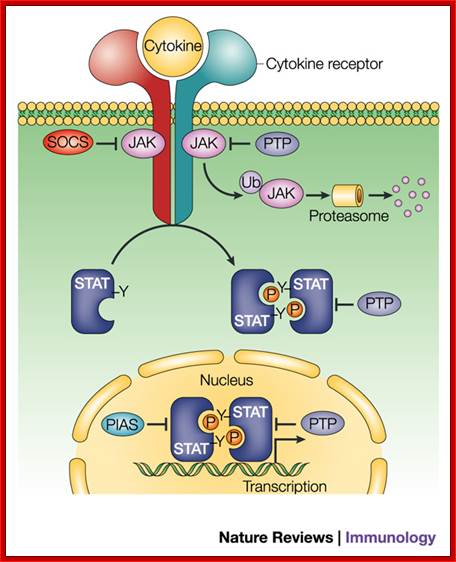
Regulation of JAK–STAT signalling in the immune system; The cytokine-activated Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway has an important role in the control of immune responses. Dysregulation of JAK–STAT signalling is associated with various immune disorders. The signalling strength, kinetics and specificity of the JAK–STAT pathway are modulated at many levels by distinct regulatory proteins. Here, we review recent studies on the regulation of the JAK–STAT pathway that will enhance our ability to design rational therapeutic strategies for immune diseases. ; http://www.nature.com/
Antigen stimulated T-helper cells, release or secrete IFN-gamma kind of Interferons or what is now called as interleukins.
Most of the other cells contain receptors for IFN-gamma.
When IFN-gamma binds to its cellular receptors and the activated cells induce transcription of a set of genes resulting in cells to be in antiviral state.
Each of the genes that respond to IFN-gamma contains, in their regulator regions, the GAF Response Elements (GAF-RE). GAF means gamma-associated factor Response Elements.
A transcriptional factor, required for IFN-gamma mediated activation, has been identified and cloned. Its Mol.wt is 91 KD and it is called STAT-1 (which is a signal transducer and activator of transcription).
IFN-gamma transduces the signal by kinase activity, which results in the phosphorylation of STAT-1 at one of the tyrosine moiety. The phosphorylated STAT-1 dimerizes and moves into the nucleus and a bind to GAF, which is already bound to GAF-RE and the gene, is activated for transcription.
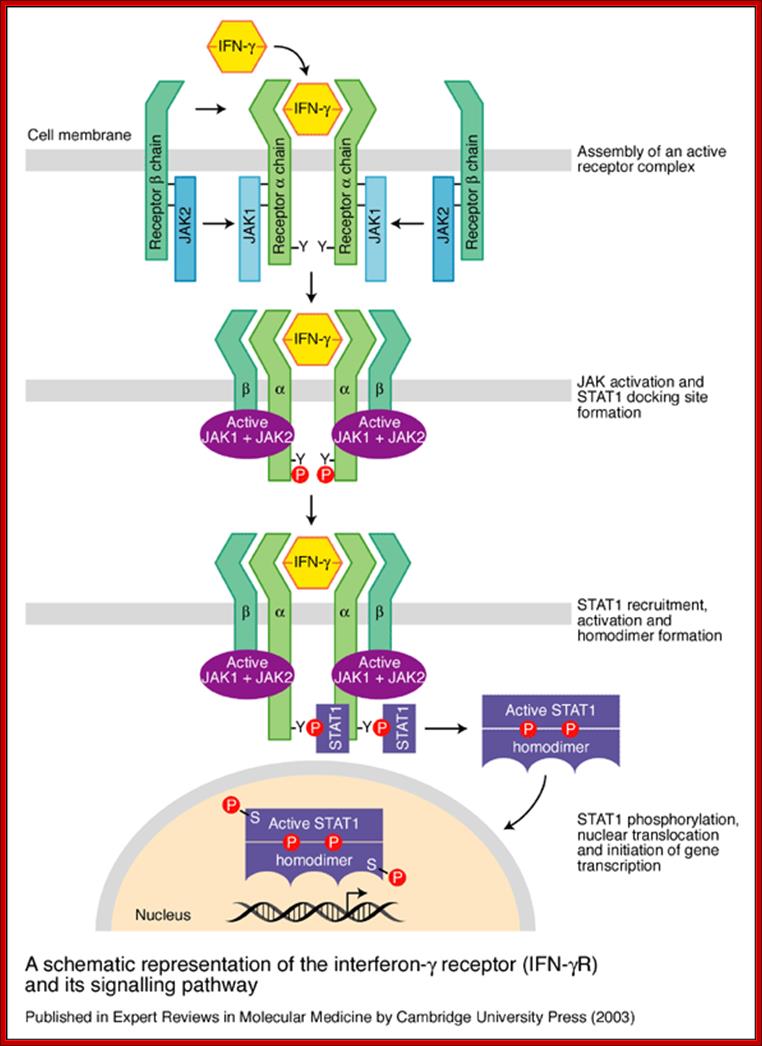
A schematic representation of the interferon-g receptor (IFN-gR) and its signalling pathway. The receptor for IFN-g has two subunits: IFN-gR1, the ligand-binding chain (also known as a chain) and IFN-gR2, the signal-transducing chain (also known as the b chain or accessory factor 1). These proteins are encoded by separate genes (IFNGR1 and IFNGR2, respectively) that are located on different chromosomes. As the ligand-binding (or a) chains interact with IFN-g they dimerizes and become associated with two signal-transducing (or b) chains. Receptor assembly leads to activation of the Janus kinases JAK1 and JAK2 and phosphorylation of a tyrosine residue on the intracellular domain of IFN-gR1. This leads to the recruitment and phosphorylation of STAT1 (for ‘signal transducers and activators of transcription’), which forms homodimers and translocates to the nucleus to activate a wide range of IFN-g-responsive genes. After signalling, the ligand-binding chains are internalized and dissociate. The chains are then recycled to the cell surface; http://www.bio.davidson.edu/
http://www.scielo.br/
Cells, mutants for STAT-1 alpha, cannot be phosphorylated because of the absence of tyrosine residues and STAT-1 Alpha does not move into the nucleus, in spite of the stimulation by IFN-gamma.
Interestingly, stimulation of cells by IFN-gamma, in the absence of IFN-alpha, leads to phosphorylation and dimerization of STAT-1 and they move into the nucleus and bind to GAF and stimulate only GAF bound to GAF response elements containing genes, it is an exclusive gene expression.
Signaling pathway of cytokine receptor families is more or less similar to interferon JAK-STAT or TYK-STAT pathways.
In a generalized version, it know known, that binding of the ligand to a receptor causes dimerization of the receptor at cytosolic side, which interact with kinases of JAK family and activates them, which in turn specifically phosphorylate a set of transcriptional factors, which then move and bind directly to response elements or bind to a factor which is already bound the response element and activate the transcriptional apparatus.
This pathway appears to operate in other hormone stimulated gene expression, ex. EGF binding to the receptor activates it to activate RTK, which then stimulate cytosolic STATs, which move into the nucleus and bind to c-Fos RE and activate genes, called serum inducible genes.
IFN induced gene expression:
Interferon alpha induced genes have specific promoters and TFs and other activator elements including enhancers.
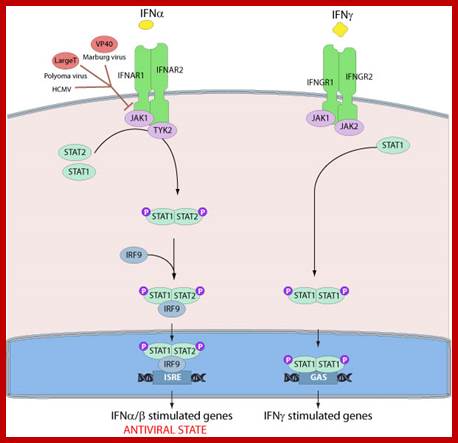
Several viruses have evolved specific mechanisms to prevent the establishment of an antiviral state by inhibiting key components of the signaling pathway. For instance, the polyomavirus large T antigen binds to Janus tyrosine kinase 1 and inactivated signaling through IFN receptors. The Marburg virus VP40 protein is able to prevent the tyrosine phosphorylation of JAK1 and subsequent phosphorylation of STAT1 and STAT2. The main pathway of type I and type III interferon induced gene expression. Binding of IFN-α to the type I interferon receptor (IFNAR1 and IFNAR2) as well as IFN-λ binding to the type III interferon receptor complex (IFN-λR1 and IL-10R2) allows the JAK kinases JAK1 and TYK2 to cross phosphorylate one another. This activates the kinases leading to phosphorylation of STAT1 and STAT2 that form a STAT1-STAT2 heterodimer. The dimer binds IRF9 forming the ISGF3 complex that migrates to the nucleus where it binds to ISRE elements thus facilitating the transcription of ISGs. http://viralzone.expasy.org
While activating the genes, the required factors bind and recruit all the cofactors and Mediator Complex MC , in such a way the DNA elements in the upstream bound by factors produce a loop, so as to interact with BTA.
Effector Mechanisms:
The interface between innate and adaptive immunity:
· Innate immunity is the initial response to microbes that prevents infections and in some cases eliminates pathogens (but pathogens have evolved mechanisms to evade)
· The effector mechanisms of innate immunity are often used to eliminate microbes even in the adaptive response (innate often the first AND the end effectors)
· Innate immunity simulates and directs adaptive responses
Comparison between Innate and Adaptive immunity:
|
Innate Immunity |
Adaptive Immunity |
|
Pathogen recognized by receptors encoded in the germline |
Pathogen recognized by receptors generated randomly |
|
Receptors have broad specificity, i.e., recognize many related molecular structures called PAMPs (pathogen-associated molecular patterns) |
Receptors have very narrow specificity; i.e., recognize a particular epitope |
|
PAMPs are essential polysaccharides and polynucleotides that differ little from one pathogen to another but are not found in the host. |
Most epitopes are derived from polypeptides (proteins) and reflect the individuality of the pathogen. |
|
Receptors are PRRs (pattern recognition receptors) |
In jawed vertebrates, the receptors are B-cell (BCR) and T-cell (TCR) receptors for antigen [Link] |
|
Immediate response |
Slow (3–5 days) response (because of the need for clones of responding cells to develop — Link) |
|
Little or no memory of prior exposure |
Memory of prior exposure [Link] |
|
Occurs in all metazoans? |
Occurs in vertebrates only |
|
Discussed on this page |
|
|
|
|
Innate and adaptive Interface: consequences of success or failure;
Activation of Dendrite and Macrophages lead to the synthesis and release of cytokines and activation leads to Lymphokines.
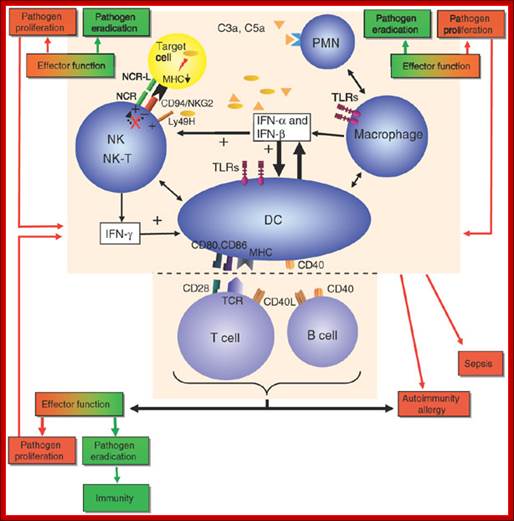
Essential to the successful removal of pathogens is the early recognition of microbes by components of the innate immune system. These involve the complement system, specialized receptors expressed on NK cells and the family of TLRs that are expressed on myeloid as well as lymphoid cells and that recognize specific microbially derived molecular structures. Successful engagement of some of these pathways leads to an inflammatory response with destruction of the pathogen alongside the enlistment of DC and T cell and/or B cell interactions. A well-orchestrated innate and adaptive immune response will lead to pathogen eradication and host immunity (green). Failure to efficiently discriminate self from non-self in innate as well as adaptive immunity can lead to pathogen proliferation and ultimately sepsis (red) and may also be the cause for development and maintenance of autoimmune diseases and allergy (red). CD40L, CD40 ligand; IFN, interferon; MHC, major histocompatibility complex; PMN, polymorphonuclear cells; TCR, T cell receptor; NCR, natural cytotoxicity receptor; NCR-L, natural cytotoxicity receptor−ligand. http://www.nature.com/
Lymphocyte secretion of Interleukins provides the interface between Innate and adaptive immunity.
Humoral Immunity:
Humoral immunity is mediated by antibodies and is the effector arm of adaptive immunity, which is responsible for defense against extracellular microbes and microbial toxins. Long lived antibodies that provide protection against infection may be produced by long lived antibody secreting cells generated by the first exposure to microbes by the first exposure to microbial antigens by activation of memory B cells by antigen.
The effector functions of antibodies include neutralization of antigens. Fc receptor–dependent phagocytoses of opsonized particles and activation of the complement system.
Antibodies block or neutralize the infectivity of microbes by the binding to microbes and sterically hindering interactions of microbes with cellular receptors. Antibodies similarly block the pathological effects of toxins to host cells.
Antibody coated (opsonized) particles are phogocytosed by the binding of Fc portions of the antibodies to phagocyte Fc receptors. There are several types of Fc receptors specific for different subclasses of IgG and for IgA and IgE antibodies and different Fc receptors bind the antibodies with varying affinities. An attachment of antigen-complexed Ig to phagocyte Fc receptors also delivers signals that stimulate the microbicidal activities of phagocytes.
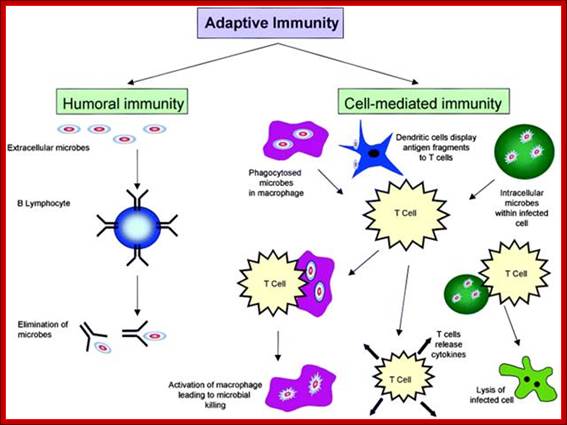
Adaptive immunity is also called specific or acquired immunity. It is activated when the innate or non-specific immune system can’t efficiently destroy the foreign organism. Specific immunity is distinguished by its specificity for an invading organism and its ability to remember an encounter so that the second time the same invader is encountered a more rapid and intense response occurs. There are two types of specific immune responses; humoral and cellular.
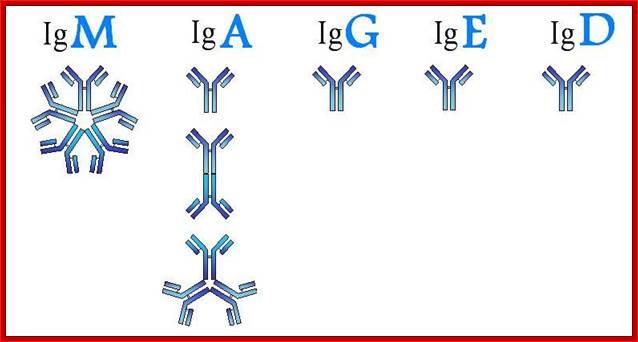
Humoral immunity is mediated by lymphocytes, which derive from stem cells in the bone marrow. Thus, they are called B cells and only those that do not attack self are released. After maturation, these B cells produce antibodies (IgG, IgM, IgA, IgE), types of proteins described in Section 5, when they encounter an antigen
http://yang-sheng.com/
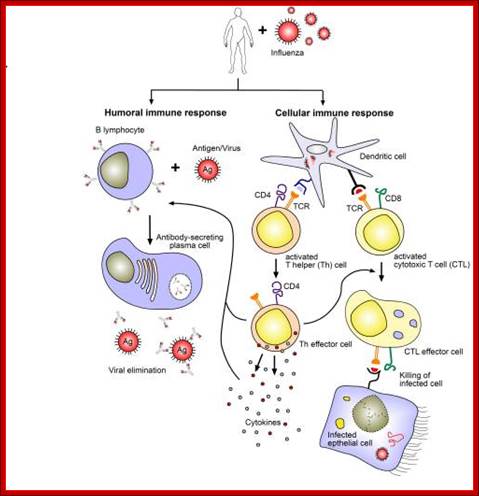
The figure shows the action of both Humoral and Cell mediated Immunoreaction; http://betournay.wikispaces.com/
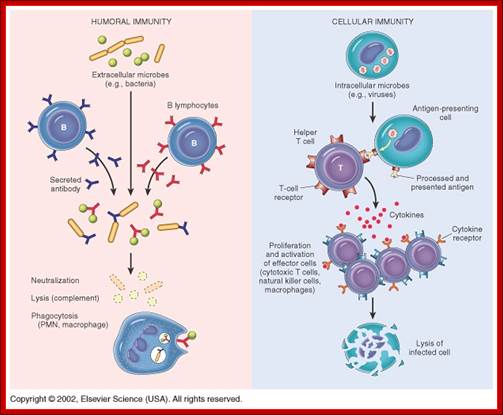
As shown in Figure , the adaptive immune response can be further broken down into cell-mediated and humoral components. Cell-mediated immunity involves the activation of macrophages and natural killer cells (destroy pathogens), antigen-specific cytotoxic T-lymphocytes (lyse infected cells), and the release of cytokines (influence functions of other cells). The humoral response refers to the production of antibodies by B-cells and activation of the accessory processes that accompany it: Th2 activation (T-helper cells), cytokine production, memory cell generation, and complement system activation. Extracellular parasites initiate the humoral immune response while intracellular parasites are susceptible to attack by the cellular response (6).;The two branches of the adaptive immune system: humoral (mediated by B lymphocyte production of antibodies) and cellular (mediated by T lymphocytes; http://betournay.wikispaces.com/
Complement System:
Historically speaking, in the late 19th century, Hans Ernst August Buchner found that blood serum contained a "factor" or "principle" capable of killing bacteria. In 1896, Jules Bordet, a young Belgian scientist in Paris at the Pasteur Institute, demonstrated that this principle had two components: one that maintained this effect after being heated, and one that lost this effect after being heated. The heat-stable component was responsible for the immunity against specific microorganisms, whereas the heat-sensitive (heat-labile) component was responsible for the non-specific antimicrobial activity conferred by all normal serum. This heat-labile component is what we now call "complement." The term "complement" was introduced by Paul Ehrlich in the late 1890s. The heat-labile antimicrobial component of fresh serum named this heat-labile component "complement," because it is something in the blood that "complements" the cells of the immune system. Ehrlich, therefore, named this heat-labile component "complement," because it is something in the blood that "complements" the cells of the immune system.
The complement system consists of serum and membrane proteins that interact in highly regulated manner to produce biologically active protein products. Two major pathways of complement activation are the alternative pathways, which is activated by the antigen and antibody complexes. The two pathways generate enzymes that cleave the C3 proteins and products of C3 become covalently attached to microbial surface or antibodies, so subsequent steps of complement activation are limited to these sites. Both pathways converge after C3 proteolysis, and the late steps consist of the proteolysis and association of several complement proteins culminating in MAC formation.
The complement system helps or “complements” the ability of antibodies and phagocytic cells to clear pathogens from an organism. It is part of the immune system called the innate immune system that is not adaptable and does not change over the course of an individual's lifetime. However, it can be recruited and brought into action by the adaptive immune system.
The complement system consists of a number of small proteins found in the blood, generally synthesized by the liver, and normally circulating as inactive precursors (pro-proteins). When stimulated by one of several triggers, proteases in the system cleave specific proteins to release cytokines and initiate an amplifying cascade of further cleavages. The end-result of this activation cascade is massive amplification of the response and activation of the cell-killing membrane attack complex. Over 25 proteins and protein fragments make up the complement system, including serum proteins, serosal proteins, and cell membrane receptors. They account for about 5% of the globulin fraction of blood serum.
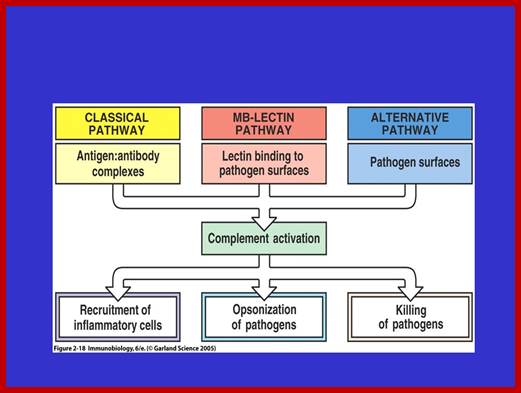
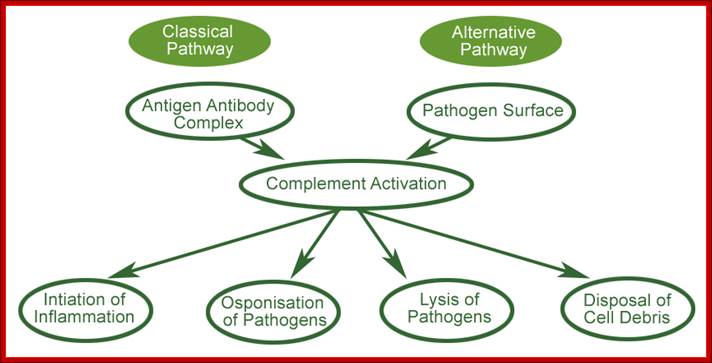
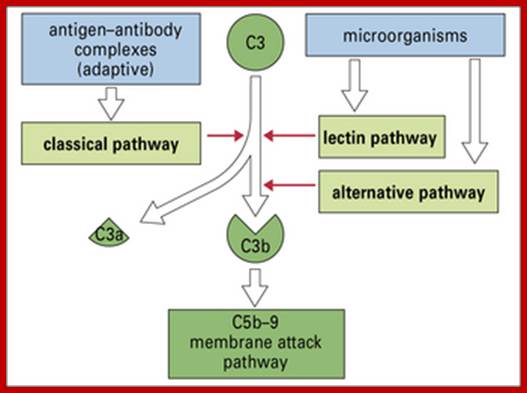
Three biochemical pathways activate the complement system: the classical complement pathway, the alternative complement pathway, and the lectin pathway. The basic functions of complement system are Opsonization, Chemotaxis, Cell lysis and Clumping of antigen bearing elements.
The proteins and glycoproteins that constitute the complement system are synthesized by the liver hepatocytes. But significant amounts are also produced by tissue macrophages, blood monocytes, and epithelial cells of the genitourinal tract and gastrointestinal tract. The three pathways of activation all generate homologous variants of the protease C3-convertase.
The classical complement pathway typically requires antigen- antibody complexes for activation (specific immune response), whereas the alternative and mannose-binding lectin pathways can be activated by C3 hydrolysis or antigens without the presence of antibodies (non-specific immune response).
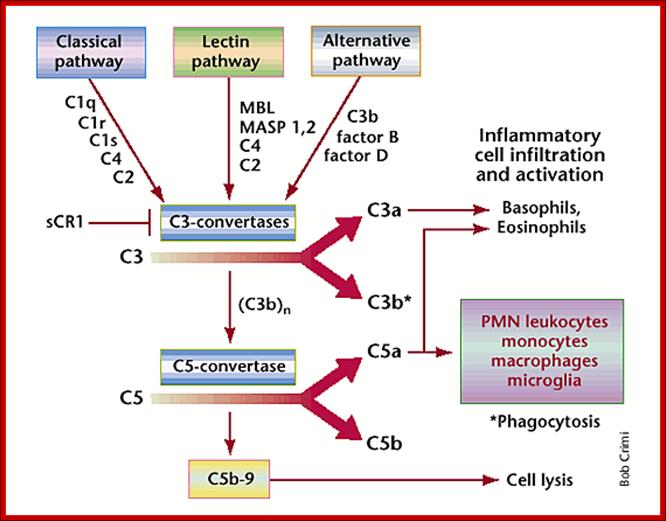
In all three pathways, C3-convertase cleaves and activates component C3, creating C3a and C3b, and causing a cascade of further cleavage and activation events. C3b binds to the surface of pathogens, leading to greater internalization by phagocytic cells by opsonization. C5a is an important chemotactic protein, helping recruit inflammatory cells. C3a is the precursor of an important cytokine (adipokine) named ASP and is usually rapidly cleaved by carboxypeptidase B. Both C3a and C5a have anaphylatoxin activity, directly triggering degranulation of mast cells as well as increasing vascular permeability and smooth muscle contraction. C5b initiates the membrane attack pathway, which results in the membrane attack complex (MAC), consisting of C5b, C6, C7, C8, and polymeric C9.
MAC is the cytolytic end product of the complement cascade; it forms a transmembrane channel, which causes osmotic lysis of the target cell. Kupffer cells and other macrophage cell types help clear complement-coated pathogens. As part of the innate immune system, elements of the complement cascade can be found in species earlier than vertebrates; most recently in the protostome horseshoe crab species, putting the origins of the system back further than was previously thought.
The defense against microbes and toxins in humans of mucosal organs is provided by IgA antibodies produced in mucosal lymphoid system and actively transported through epithelial cells into the lumen.
A protective immunity neonate is a form of passive immunity provided by maternal antibodies transported across placenta or ingested and transported across the gut epithelium by specialized neonatal Fc receptors.
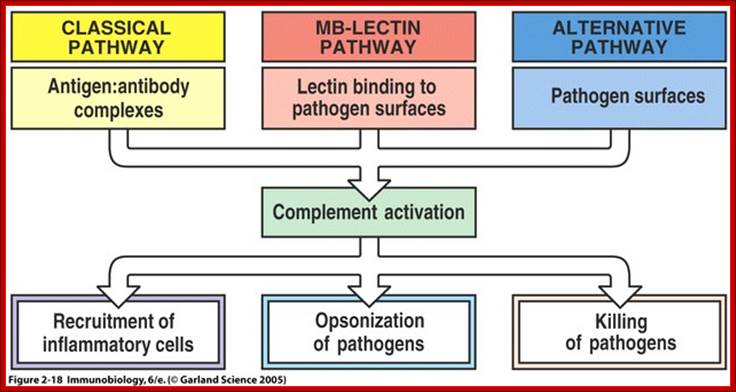
The main functions of complement are recruitment of inflammatory cells, opsonization of pathogens, and killing of pathogens ; Once MBL recognizes a pathogen, its lectin domain will bind to mannose, or other carbohydrate sugar residues on the pathogen surface, and activate complement via the MB-lectin pathway ; http://www.bio.davidson.edu/
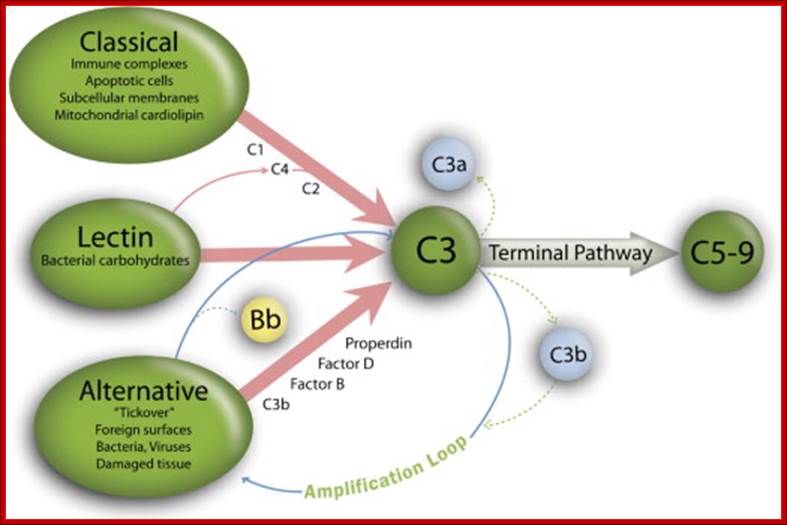
Figure: Classical, lectin, and alternative complement pathways. The 3 complement pathways converge to generate C3 convertase that cleave C3, creating C3a and C3b. C3b is a major effector molecule of the complement system. C3b binds covalently to the surface of nearby cells, opsonizes the cells for phagocytosis, and initiates the reactions that lead to the formation of the membrane attack complex (C5-9). Bystander host cells are protected from excessive complement activation by cell-bound and circulating complement regulatory proteins. The alternative pathway also has the unique ability to serve as an amplification system for the classical and lectin pathways. In this study, we examined the complement activation fragment C3a, which can arise from activation of any of the 3 complement pathways and also Bb. The Bb activation fragment is primarily associated with alternative complement pathway activation but can also arise as a result of activation of initiation of the complement cascade by the classical or lectin pathways through the alternative complement pathway amplification loop. Bb is a protease generated during the sequential activation of both the alternative pathway and the activation loop and is required for further steps of the pathway. The alternative pathway and amplification loop are reviewed in detail elsewhere. Lynch. The complement system, obesity, and preeclampsia. Am J Obstet Gynecol 2012. http://www.ajog.org
The Top figures show the three pathways, the above figure shows the mechanism by which Payers Patch-complement system acts; http://iahealth.net/
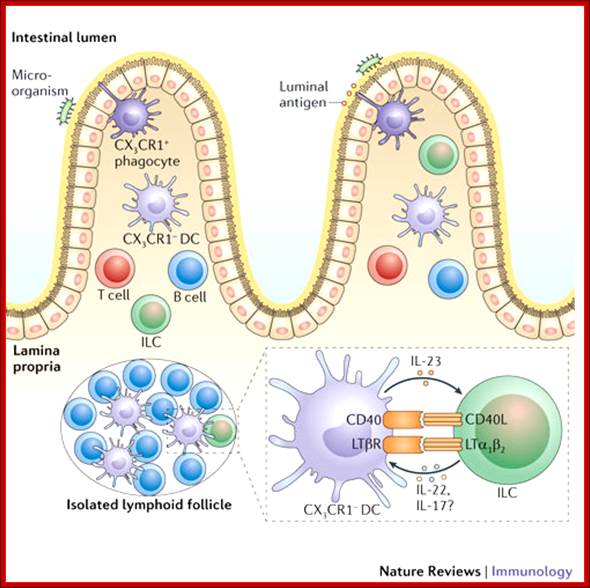
TNF superfamily members and immune responses in the intestine; In the small intestine, dendritic cells (DCs) are present in the organized Peyer's patches (not shown), in isolated lymphoid follicles and in the lamina propria. DCs express different cell-surface receptors depending on their location in the small intestine. DCs can interact with innate lymphoid cells (ILCs) to provoke ILC activation. DC-expressed members of the tumor necrosis factor (TNF) superfamily may be important for promoting the expression of interleukin-23 (IL-23), which provokes the production of IL-22 by ILCs to protect the host from infection. This form of crosstalk is reminiscent of the crosstalk that occurs in the periphery during DC licensing. CD40L, CD40 ligand; CX3CR1, CX3C-chemokine receptor 1; LTα1β2, lymphotoxin-α1β2; LTβR, LTβ receptor. Leslie Summers deLuca & Jennifer L. Gommerman; http://www.nature.com/
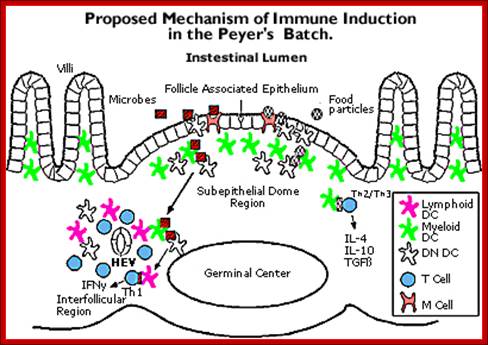
The above figure shows the mechanism by which Payers Patch-complement system act; www.repairnwiring.com/Peyers-Patch-Diagram.html
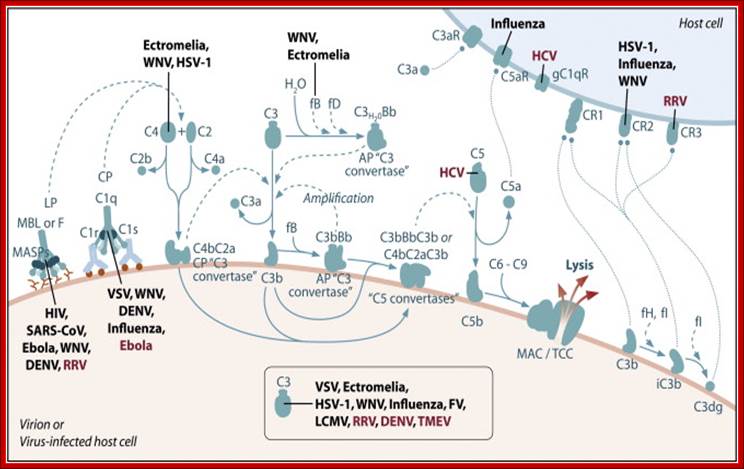
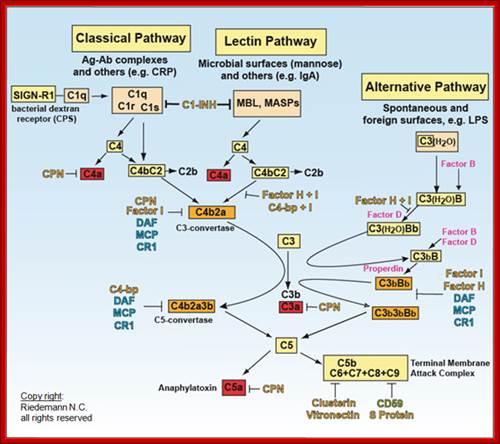
Schematic of the complement system. Complement is activated by three major pathways. The classical pathway is primarily activated when C1q interacts with IgM and certain IgG isotypes bound to antigen. C1q-associated C1s cleaves C4 and C2 to form the classical pathway (CP) C3 convertase (C4bC2a). The lectin pathway (LP) is initiated by carbohydrate pattern recognition receptors such as mannose-binding lectin (MBL) and the ficolins (F) which are in a complex with enzymes known as MBL-associated serine proteases (MASPs). MASP-2 activates the complement system by cleaving both C4 and C2 to form the C4bC2a C3 convertase. The alternative pathway (AP) is activated by spontaneous hydrolysis of C3 (C3-H2O). This pathway also functions as an amplification loop for the cleavage of C3 initially triggered by other mechanisms. C3-H2O or C3b bound to target surfaces are bound by the protease factor B (fB). Factor D (fD) is a serine protease that cleaves C3-H2O or C3b-bound fB, resulting in the generation of Bb and formation of the alternative pathway C3 convertase (C3bBb). Both the classical and alternative convertases function to cleave C3 to C3a and C3b. Cleavage of C3 exposes a reactive thioester bond in C3b that allows for the covalent attachment of C3b to target surfaces. In addition, C3b can bind to either the classical or alternative C3 convertases resulting in a change of the substrate specificity of the convertases from C3 to C5. These C5 convertases cleave C5 to C5a and C5b. Release of C5b promotes assembly of the C5b–C9 membrane attack complex (MAC) which can directly lyse pathogens or pathogen-infected cells. C3b is further cleaved by factor I (fI), a function enhanced by factor H (fH), to generate degradation products such as iC3b and C3dg. C3b and its degradation products interact with cellular receptors to regulate effector functions such as phagocytosis and B cell activation. The anaphylatoxins C3a and C5a interact with specific receptors to promote chemotaxis and regulate effector functions of cells of both the innate and adaptive immune response.
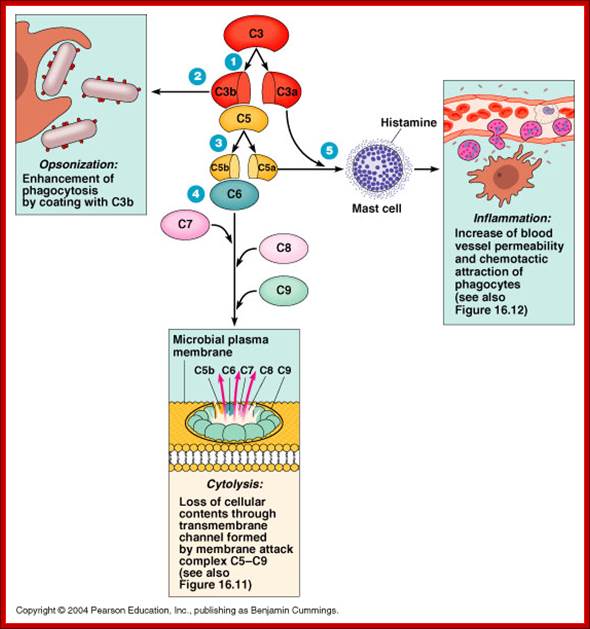
Cleaving of. C3 b binds to bacteria and leads to Opsonization. C3a interacts with mast cells leading to the release of histamines. This increase blood vessel permeability and Phagocytes get attracted.. The C3b and C5 interaction leads yo cleavage of C5 C5a and C5b. The C5b contacts C6, this leads to a cascade of assembly of C factors (C5b, C6, C7, C8 and C9) and the same are transported to membranes and create a hole in the membrane and release the components-lysis. http://slideplayer.com
The classical pathway is triggered by activation of the C1-complex. The C1-complex is composed of 1 molecule of C1q, 2 molecules of C1r and 2 molecules of C1s, or C1qr2s2. This occurs when C1q binds to IgM or IgG complexed with antigens. A single IgM can initiate the pathway, while multiple IgGs are needed. This also occurs when C1q binds directly to the surface of the pathogen. Such binding leads to conformational changes in the C1q molecule, which leads to the activation of two C1r molecules. C1r is a serine protease. They then cleave C1s (another serine protease). The C1r2s2 component now splits C4 and then C2, producing C4a, C4b, C2a, and C2b. C4b and C2a bind to form the classical pathway C3-convertase (C4b2a complex), which promotes cleavage of C3 into C3a and C3b; C3b later joins with C4b2a (the C3 convertase) to make C5 convertase (C4b2a3b complex). The inhibition of C1r and C1s is controlled by C1-inhibitor. C3-convertase can be inhibited by Decay accelerating factor (DAF), which is bound to erythrocyte plasma membranes via a GPI anchor. Paroxysmal nocturnal hemoglobinuria is caused by complement breakdown of RBCs due to an inability to make GPI. Thus the RBCs are not protected by GPI anchored proteins such as DAF.
Alternative pathway:
Main article: Alternative complement pathway
The alternative pathway is continuously activated at a low level, analogous to a car engine at idle, as a result of spontaneous C3 hydrolysis due to the breakdown of the internal thioester bond(C3 is mildly unstable in aqueous environment). The alternative pathway does not rely on pathogen-binding antibodies like the other pathways. C3b that is generated from C3 by a C3 convertase enzyme complex in the fluid phase is rapidly inactivated by factor H and factor I, as is the C3b-like C3 that is the product of spontaneous cleavage of the internal thioester. In contrast, when the internal thioester of C3 reacts with a hydroxyl or amino group of a molecule on the surface of a cell or pathogen, the C3b that is now covalently bound to the surface is protected from factor H-mediated inactivation. The surface-bound C3b may now bind factor B to form C3bB. This complex in the presence of factor D will be cleaved into Ba and Bb. Bb will remain associated with C3b to form C3bBb, which is the alternative pathway C3 convertase. The C3bBb complex is stabilized by binding oligomers of factor P. The stabilized C3 convertase, C3bBbP, then acts enzymatically to cleave much more C3, some of which becomes covalently attached to the same surface as C3b. This newly-bound C3b recruits more B,D and P activity and greatly amplifies the complement activation. When complement is activated on a cell surface, the activation is limited by endogenous complement regulatory proteins, which include CD35, CD46, CD55 and CD59, depending on the cell. Pathogens, in general, don't have complement regulatory proteins (there are many exceptions, which reflect adaptation of microbial pathogens to vertebrate immune defenses). Thus, the alternative complement pathway is able to distinguish self from non-self on the basis of the surface expression of complement regulatory proteins. Host cells don't accumulate cell surface C3b (and the proteolytic fragment of C3b called iC3b) because this is prevented by the complement regulatory proteins, while foreign cells, pathogens and abnormal surfaces may be heavily decorated with C3b and iC3b. Accordingly, the alternative complement pathway is one element of innate immunity.
Once the alternative C3 convertase enzyme is formed on a pathogen or cell surface, it may bind covalently another C3b, to form C3bBbC3bP, the C5 convertase. This enzyme then cleaves C5 to C5a, a potent anaphylatoxin, and C5b. The C5b then recruits and assembles C6, C7, C8 and multiple C9 molecules to assemble the membrane attack complex. This creates a hole or pore in the membrane that can kill or damage the pathogen or cell.
Lectin pathway:
Main article: Lectin pathway
The lectin pathway is homologous to the classical pathway, but with the opsonin, mannose-binding lectin (MBL), and ficolins, instead of C1q. This pathway is activated by binding mannose-binding lectin to mannose residues on the pathogen surface, which activates the MBL-associated serine proteases, MASP-1, and MASP-2 (very similar to C1r and C1s, respectively), which can then split C4 into C4a and C4b and C2 into C2a and C2b. C4b and C2a then bind together to form C3-convertase, as in the classical pathway. Ficolins are homologous to MBL and function via MASP in a similar way. In invertebrates without an adaptive immune system, ficolins are expanded and their binding specificities diversified to compensate for the lack of pathogen-specific recognition molecules.
In stroke, complement will get you nowhere; Early components in the classical, lectin or alternative pathways recognize injured cells and initiate a cascade leading to inflammatory cell infiltration and activation, and destruction of the damaged cells. After C1q, mannose binding ligand (MBL) or C3b recognize and bind an injured cell, a proteolytic signaling cascade is initiated. This cascade leads to generation of C3 convertases that activate cleavage of C3 into C3a and C3b, generating C5 convertase. C5 convertase activates cleavage of C5 into C5a and C5b. C3a and C5a attract basophils and eosinophils to damaged tissue. C5a also induces infiltration of PMN leukocytes, monocytes and macrophages to sites of injury. In the central nervous system, microglia may be similarly activated by complement. Generation of the attack complex C5b-9 is also responsible for lytis of selected cell populations, such as bacteria and other cells.; http://www.nature.com/
The Model of complement cascade
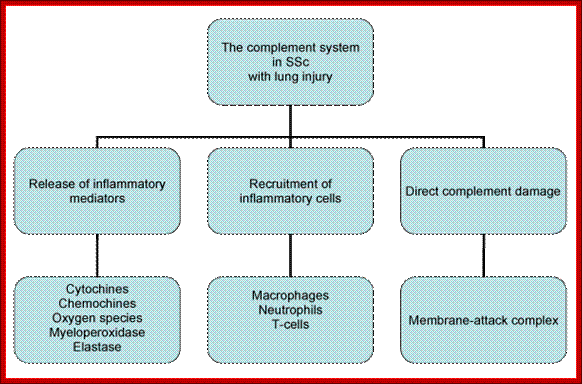
Figure: Role of complement system in SSc with lung involvement. Complement bioactive molecules, such as C3a and C5a anaphylatoxins; trigger a series of events that culminate in the recruitment of phagocytic cells, release of inflammatory molecules, activation of T-cell and augmentation of antibody response at the site of inflammation. The membrane-attack complex (MAC), resulting from distal complement component activation, can directly contribute to tissue damage through the lysis of cells, that are not properly protected by complement regulators. https://www.omicsonline.org
Complemt/Clinical gate; https://clinicalgate.com
Scheme showing the cascade of events during the activation of the complement system.
Nine complement components in the classical pathway are designated by a capital letter C and numbers 1-9. Two proteins that participate in the alternate pathway are termed factors and are represented by capital letters B and D. Proteolytically cleaved components of proteins are expressed by lowercase letters (eg, C2a, C2b). Inactive components are designated with an "i" (eg, inactivated C3b is termed iC3b). The proteins work with our immune system and play a role in the development of inflammation. There are nine major complement proteins. They are labeled C1 through C9. This test measures C3.
Regulation of the complement system
The complement system has the potential to be extremely damaging to host tissues, meaning its activation must be tightly regulated. The complement system is regulated by complement control proteins, which are present at a higher concentration in the blood plasma than the complement proteins themselves. Some complement control proteins are present on the membranes of self-cells preventing them from being targeted by complement. One example is CD59, also known as protectin, which inhibits C9 polymerization during the formation of the membrane attack complex. The classical pathway is inhibited by C1-inhibitor, which binds to C1 to prevent its activation.
The Humoral Immune system:
B-cells (so-called because they mature in the bone marrow) are white blood cells that work chiefly by producing soluble substances known as antibodies. Each B cell is programmed to make one specific antibody. When a B cell encounters its specific or eliciting antigen (along with various accessory cells), it differentiates into a plasma cell. Each of these cells once activated undergo differentiation and some produce one kind of Igs and some undergo differentiation into memory cells.
B Cells and MS: understanding their scientific impact; ;B cell; http://www.roche.com
B cell (electronic micrograph) and a B cell covered with Bacteria. http://www.insanmucizesi.com
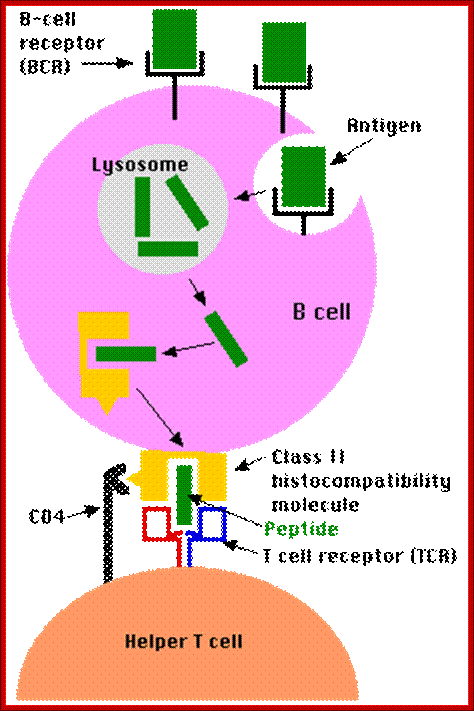
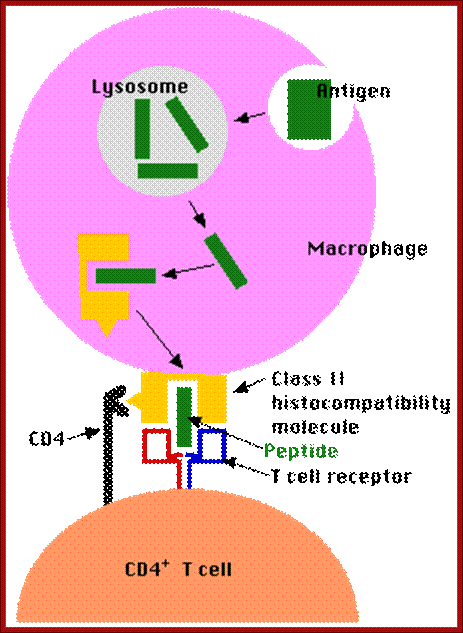
B cells are also antigen-presenting cells — Link. They bind antigen with their BCRs and engulf it into lysosomes. They then transport the digested fragments to the cell surface incorporated in class II histocompatibility molecules just as macrophages and dendritic cells do.
Pathogens coated with fragments of the complement protein C3 are not only opsonized for phagocytosis but also bind more strongly to B cells that have bound the pathogen through their BCR. This synergistic effect enables antibody production to occur at doses of antigen far lower than would otherwise be needed.
B-Cells are not only produced in the bone marrow but also mature there. Each B-Cell is specific for a particular antigen. The specificity of binding resides in the BCR (B-Cell receptor) for antigen. They are integral membrane proteins. They are present in thousands of identical copies exposed at the cell surface. They are made before the cell ever encounters an antigen.
http://www.phartoonz.com/
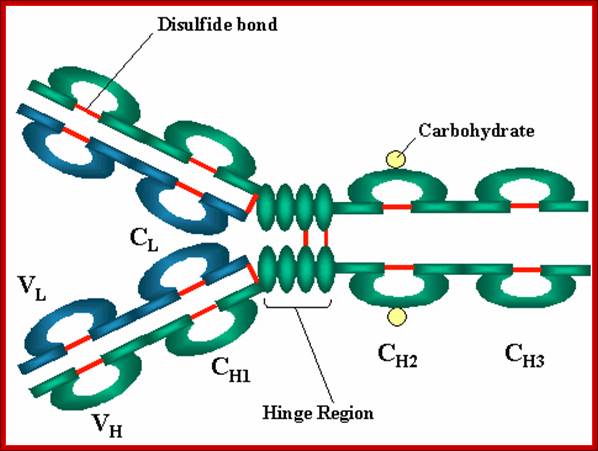
Basic structure- A Heavy and light chains, B. Disulfide bonds, C. Variable V and Constant C regions, D. Hinge region, E. Domain b light chain V/Ll and C/L, heavy chain domains; V/H, C/H, CH3 or CH4, F. Oligosaccharides; Variable regions: Comparisons of the amino acid sequences of the variable regions of immunoglobulin’s show that most of the variability resides in three regions called the hyper variable regions or the complementarity determining regions as illustrated in figure 3. Antibodies with different specificities (i.e. different combining sites) have different complementarity determining regions while antibodies of the exact same specificity have identical complementarity determining regions (i.e. CDR is the antibody combining site). Complementarity determining regions are found in both the H and the L chains. http://pathmicro.med.sc.edu/
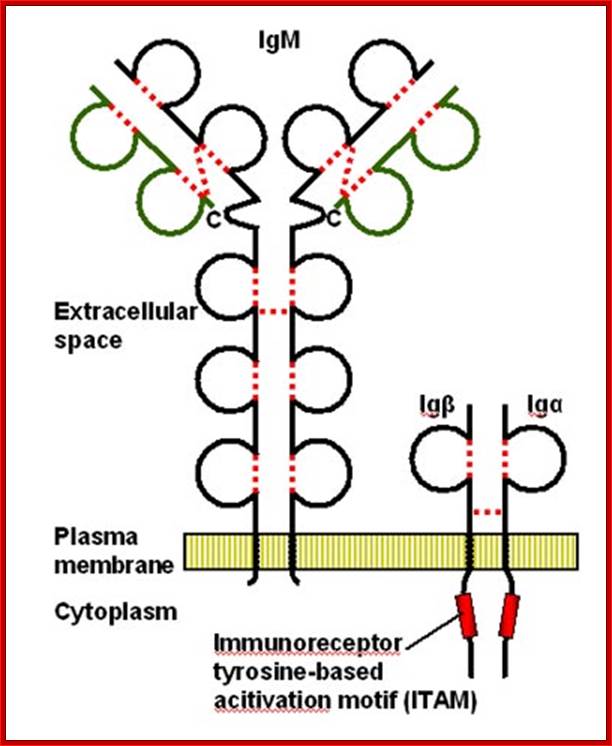
Cell membrane IgM on the surface of mature B cell is associated with Igα and Igβ molecules. The resulting complex constitutes the B cell antigen receptor complex. Igα and Igβ contain ITAMs in their cytoplasmic tails that mediate signal transduction. http://www.roswellpark.edu/
B-Cell receptor complex usually consist of an antigen-binding subunit (the membrane immunoglobulin or MIg), which is composed of two IgHs (Immunoglobulin Heavy Chains) and two IgLs (Immunoglobulin Light Chains), and a signaling subunit, which is a disulfide-linked heterodimer of Ig-Alpha (CD79A) and Ig-Beta (CD79B) proteins. During the differentiation of the B-Cell (and long before any possible encounter with an antigen), the DNA in this locus is cut and recombined to make an intact gene for the heavy chain. This gene can then be transcribed into mRNA, which is, in turn, translated into the heavy polypeptide chain. All isotypes of mIg have very short cytoplasmic tails. Both mIgM and mIgD have a cytoplasmic domain, which are only 3 amino acids in length. The cytoplasmic tails of mIg are too short to be able to associate with intracellular signaling molecules.
Since mIg is always associated with the Ig-alpha/Ig-beta heterodimer collectively forming B-Cell receptor complex (BCR), two molecules of this heterodimer associate with one mIg to form a single BCR.
The Ig-alpha/Ig-beta heterodimer carries out the signal transducing function of the complex. The Ig-alpha chain has a long cytoplasmic domain containing 61 amino acids while the Ig-beta chain has a long cytoplasmic domain containing 48 amino acids.
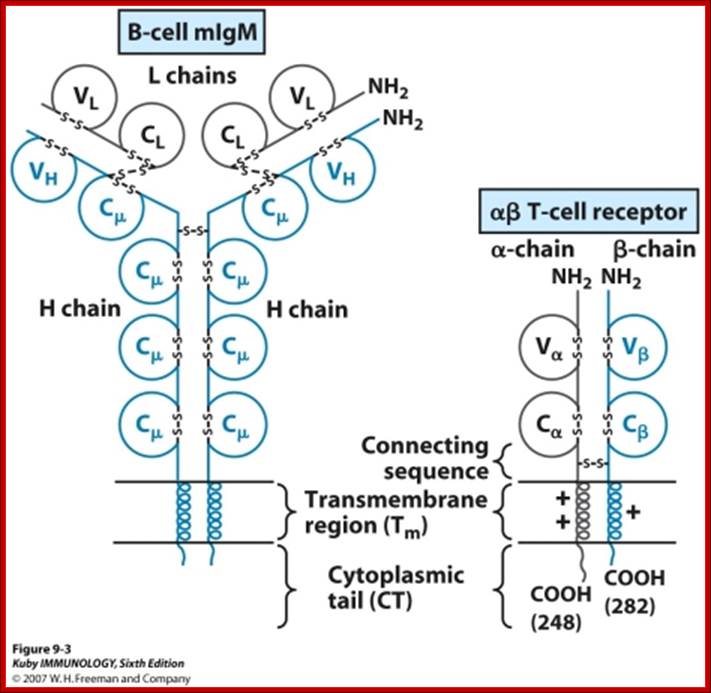
BCR have a unique binding site. This site binds to a portion of the antigen called an antigenic determinant or epitope. The binding depends on complementarity of the surface of the receptor and the surface of the epitope. The binding occurs by non-covalent forces. In the absence of specific antigen, mature B-Cells survive in the peripheral circulation for only a few days. Cells which do not encounter antigen within this period of time undergo apoptosis. This is necessary in order to maintain an optimal circulation of B-lymphocytes in the peripheral circulation. When the receptor is on the cell surface of B-lymphocytes it functions to transmit intracellular signals that regulate cell growth and differentiation and it binds to antigen for the generation of the immune response. http://www.imgt.org/IMGTeducation
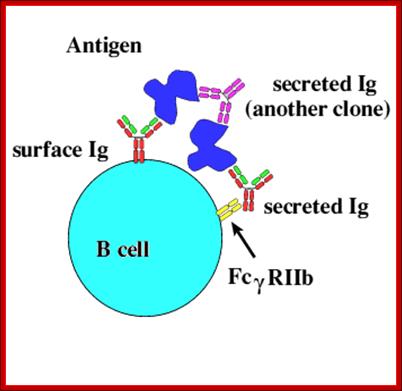
B cell IgRact as receptor and bind to antienes which can be bound by secreted Igs. https://ehumanbiofield.wikispaces.com
Most B-Cell antigens are T dependent. In other words, the B-Cell requires direct contact by TH lymphocytes as well as exposure to TH lymphocyte cytokines in order to be fully activated.
The best known T independent antigen is bacterial cell wall lipopolysaccharide, flagillin which stimulates specific Igs at high concentration and the Igs are polyclonal, and B cells proliferate and produce memory cells. The cell bound receptors are but Igs called IgM.
The B1 cells are produced in fetus, and B2 cells are produced after birth. They produce antibodies and they act as antigen presenting cells (APC); they don’t develop into memory cells. In January 2011 human B1 cells were found to have marker profile of CD20+CD27+CD43+CD70- and could either be CD5+ or CD5-.[1] CD5-CD72 is thought to mediate B cell-B cell interaction. B1 cells express IgMs in large numbers and have poly-epitope facility.
The bound antigen is engulfed by endocytosis and digested into fragment, and then they are displayed on the surface of the B cell PM complexed with MHCII proteins. The helper CD4 T cells with their TCR, they are called helper Th-Cells.
Antigen presentation: http://users.rcn.com/ /jkimball.ma.ultranet
Antigen presentation: http://users.rcn.com/ /jkimball.ma.ultranet
Antigen presentation; http://users.rcn.com/ /jkimball.ma.ultranet

What are B cells, Basic immunology concepts Review resources; Read,
Steps in production of antibodies by B cells: 1. Antigen is recognized and engulfed by B cell 2. Antigen is processed 3. Processed antigen is presented on B cell surface 4. B cell and T cell mutually activate each other 5. B cells differentiate into plasma cells to produce soluble antibodies. http://antibodycells.com/
Humoral Immunity:
The term "humoral" refers to the non-cellular components of the blood, such as plasma and lymphatic fluid. The humoral immune response denotes immunologic responses that are mediated by antibodies. However, both B and T lymphocytes, as well as dendritic cells and other antigen presenting cells, are necessary for the formation of antigen-specific antibody.
Humoral immunity includes the primary and secondary immune responses to antigen. During the primary immune response, an antigen is encountered by the host for the first time. Virgin B cells need to be activated and proliferate before an effective immune response can be generated. This primary response may be too slow to protect against many pathogens, therefore polyspecific natural antibodies with low affinity and the innate immune system may be utilized to limit microbial replication at the onset of infection. By comparison, the secondary antibody response, which results from the activation of a memory B cell, is faster and more effective in halting the progress of infection due to increased antibody binding affinities.
Vaccination induces a primary immune response so that the patient produces the faster and more effective secondary response upon natural exposure to a pathogen, and is one of the most important contributions of immunology to disease prevention.
An overview of the humoral immune response will be provided here. Discussions of immunoglobulin structure, function, and genetics, as well as a review of B cell development are found separately. (See "Function and clinical applications of immunoglobulins" and "Immunoglobulin genetics" and "Normal B and T lymphocyte development".)
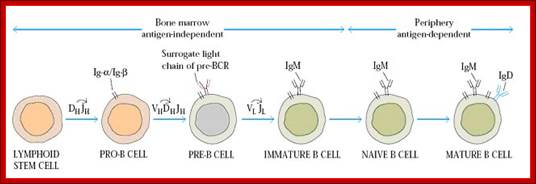
Once stem cells programme for B cell, the progenitor cell develops in temporal fashin and stage by stages, such as pre-pro, early pro, late Pro large Pre-B and small pre-B , immature B and finally mature B cells. When B cells fails to in any of the steps of processing it is subjected apoptosis.
|
Stage |
Ig |
CD19? |
|||
|
Progenitor (or pre-pro) B cells |
germline |
- |
Yes |
No |
|
|
Early Pro (or pre-pre)-B cells |
undergoes D-J rearrangement |
germline |
- |
Yes |
No |
|
Late Pro (or pre-pre)-B cells |
undergoes V-DJ rearrangement |
germline |
- |
Yes |
|
|
Large Pre-B cells |
is VDJ rearranged |
germline |
IgM in cytoplasm and surface |
Yes |
|
|
Small Pre-B cells |
is VDJ rearranged |
undergoes V-J rearrangement |
IgM in cytoplasm and surface |
Yes |
Yes |
|
Immature B cells |
is VDJ rearranged |
VJ rearranged |
IgM on surface |
No |
Yes |
|
Mature B cells |
is VDJ rearranged |
VJ rearranged |
No |
Yes |
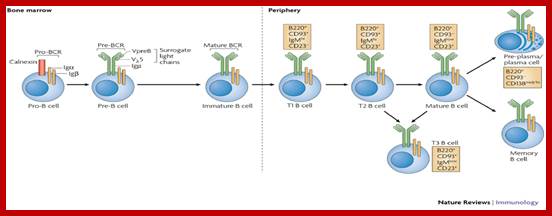
Stages of B-cell developmentB-cell development occurs in both the
bone marrow and peripheral lymphoid tissues such as the spleen. In the bone
marrow, development progresses through the pro-B-cell, pre-B-cell and
immature-B-cell stages. During this differentiation, rearrangements at the
immunoglobulin locus result in the generation and surface expression of the
pre-B-cell receptor (pre-BCR, which is comprised of an Ig![]() heavy chain and surrogate light chains (VpreB or V
heavy chain and surrogate light chains (VpreB or V![]() 5)) and finally a mature BCR (comprised of rearranged heavy-
and light-chain genes) that is capable of binding antigen. At this immature
stage of development, B cells undergo a selection process to prevent any further
development of self-reactive cells. Both receptor editing and clonal deletion
have a role at this stage. Cells successfully completing this checkpoint leave
the bone marrow as transitional B cells, eventually maturing into mature
follicular B cells (or marginal-zone B cells). Following an immune response,
antigen-specific B cells develop into either plasma (antibody-secreting) cells
or memory B cells. Transitional 3
(T3) B cells, once thought to be part of the linear development from immature
to mature B cells, are now thought to represent primarily self-reactive anergic
B cells (also known as An1 B cells). B-cell anergy: from transgenic models to naturally
occurring anergic B cells?
5)) and finally a mature BCR (comprised of rearranged heavy-
and light-chain genes) that is capable of binding antigen. At this immature
stage of development, B cells undergo a selection process to prevent any further
development of self-reactive cells. Both receptor editing and clonal deletion
have a role at this stage. Cells successfully completing this checkpoint leave
the bone marrow as transitional B cells, eventually maturing into mature
follicular B cells (or marginal-zone B cells). Following an immune response,
antigen-specific B cells develop into either plasma (antibody-secreting) cells
or memory B cells. Transitional 3
(T3) B cells, once thought to be part of the linear development from immature
to mature B cells, are now thought to represent primarily self-reactive anergic
B cells (also known as An1 B cells). B-cell anergy: from transgenic models to naturally
occurring anergic B cells?
John C. Cambier, Stephen B. Gauld, Kevin T. Merrell & Barbara J. Vilen; http://www.nature.com
B lymphocyte has different names like Pre-B cell, Immature B cell, mature B cell, Naive B cell, effector B cell, Plasma cell and memory B cell. These names provided to B cells at different period of their life time and place of occurrence.
During B cell development an important process known as DNA rearrangement occurs for Ig genes, at this stage Pre-B cell is called as immature B cell. After rearrangement completed, genes start to express and B Cell Receptor (BCR) appears on the membrane of immature B cell because BCR is mainly of immunoglobins. At this stage immature B cell said to be mature B cell. They possess well developed BCR which is made up of membrane bound Ig M and Ig D. Thus B cell development and maturation of B cell occurs in primary lymphoid organ Bone marrow. These mature B cells then start to move towards secondary or peripheral lymphoid organs to encounter antigen. During their transport, they are known as naive B cells i.e. they are not exposed to antigen.
After activation, B cell undergoes proliferation. After proliferation, with the help of cytokines, they undergo differentiation process through which two types of cells produced namely effector B cell (Plasma cell) and memory B cell. Plasma cell is the type of B cell which can secrete antibodies depending upon the cytokines and class switching process. All plasma cell initially produce Ig M type and latter they switched to produce either Ig G or Ig A or Ig E. They are also actively involved in primary immune response i.e. present infection clearance reaction. Memory B cell does not produce secretary antibodies but they have membrane bound immunoglobins which is used for antigen recognition in the future. They place vital role in secondary immune response. Once B cell differentiated to effector B cell and memory B cell, they can move to tertiary lymphoid organ for their action.
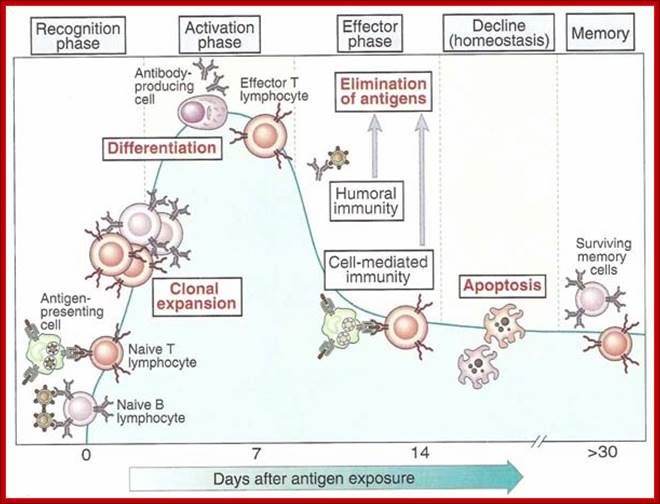
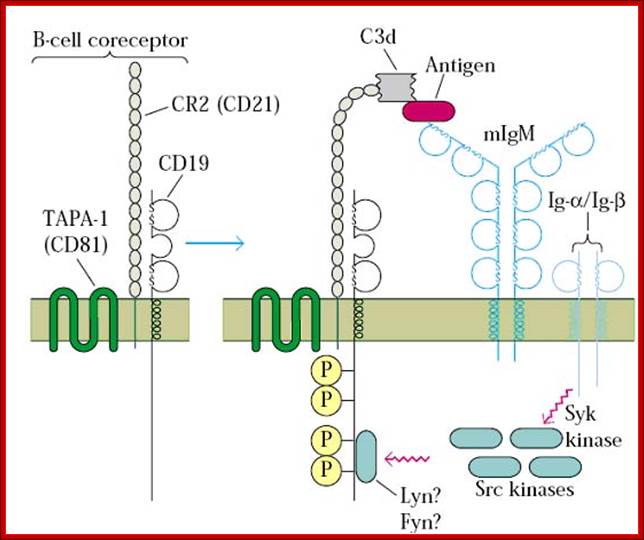
B cell receptors and coreceptors; biosiva.50webs.org/plasmacell.htm
Follicular B cells (FO B cells) From Wikipedia, the free encyclopedia
The FO B cells are a type of B cell that resides in primary and secondary lymphoid follicles (containing germinal centers) of secondary and tertiary lymphoid organs, including spleen and lymph nodes.
The mature B cells from the spleen can be divided into two main populations: the FO B cells, which constitute the majority, and the marginal zone B-cells, lining outside the marginal sinus and border the red pulp. FO B cells express high levels of IgM, IgD, and CD23; lower CD21; and no CD1 or CD5, readily distinguishing this compartment from B1 B cells and marginal zone B-cells
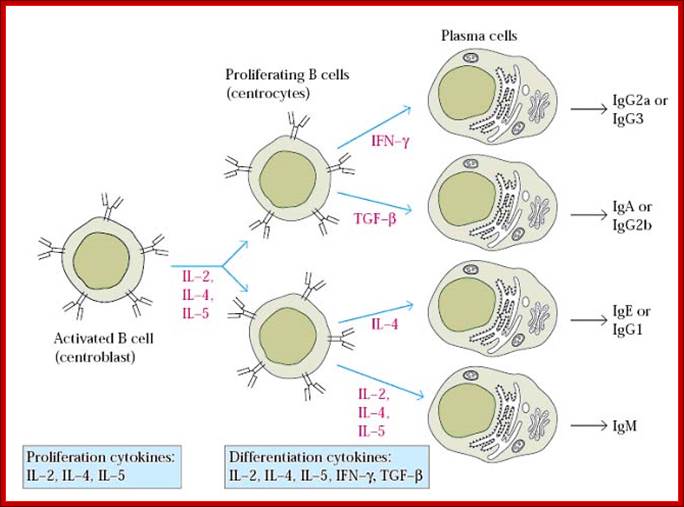
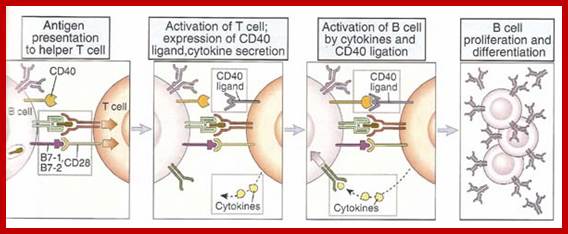
B cell activation by T cell:
After antigen presented by B cell, T cells express CD40 ligand (CD40L) on its surface and release cytokines. CD40L is a T cell membrane protein that is structurally homologous to TNF and Fas ligand. CD40L interacts with CD40 on B cell when T cell and B cell binds. CD40L binding to CD40 results in oligomerization of CD40 molecules and this induces the association of cytoplasmic proteins called TNF receptor associated factors (TRAFs) to the cytoplasmic domain of CD40. The TRAFs recruited to CD40 initiate enzyme cascades that lead to the activation and nuclear translocation of transcription factors, including NF-kb and AP-1. Activated helper T cells were secret cytokines that act in concert with CD40L to stimulate B cell proliferation and production of antibodies of different isotypes. ; http://biosiva.50webs.org
Marginal zone B-cell
From Wikipedia, the free encyclopedia
Marginal zone B cells are noncirculating mature B cells that segregate anatomically into the marginal zone (MZ) of the spleen.[1] This region contains multiple subtypes of macrophages, dendritic cells, and the MZ B cells; it is not fully formed until 2 to 3 weeks after birth in rodents and 1 to 2 years in humans.[2] The MZ B cells within this region typically express high levels of sIgM, CD21, CD1, CD9 with low to negligible levels of sIgD, CD23, CD5, and CD11b that help to distinguish them phenotypically from FO B cells and B1 B cells.
Similar to B1 B cells, MZ B cells can be rapidly recruited into the early adaptive immune responses in a T cell independent manner. The MZ B cells are especially well positioned as a first line of defense against systemic blood-borne antigens that enter the circulation and become trapped in the spleen. It is believed they are especially reactive to bacterial cell wall components and self-antigens which are the products of aging. MZ B cells also display a lower activation threshold than their FO B cell counterparts with heightened propensity for PC differentiation that contributes further to the accelerated primary antibody response.
Antibody-Antigen Interactions
An antigen is usually defined as a substance that causes an immune response when introduced into an organism and is capable of binding with the specific antibodies. This binding is mediated by the sum of many weak interactions between the antigen and antibody. These weak interactions include hydrogen bonds, van der Waals forces, and ionic and/or hydrophobic interactions.
These interactions can only take place if the antigen and antibody molecules are close enough for some of the individual atoms to fit into complementary recesses. The complementary regions of an antibody are its 2 antigen binding sites (thus the antibody is said to be bivalent). The corresponding region(s) of the antigen is referred to as an antigenic determinant. Most antigens have multiple determinants; if 2 or more are identical, the antigen is said to be multivalent.
As the binding of an antibody (ab) to its antigen (ag) is reversible, the binding reaction can be expressed as:
ab + ag ↔ ab:ag
The strength of the interaction is expressed as the affinity constant Ka, where:
Ka = [ab:ag]/[ab][ag]
In this equation, [ab:ag] is the molar concentration of the antibody-antigen complex, and [ab] and [ag] are the molar concentrations of the antibody and antigen, respectively. Affinity constants can vary widely between different antibodies and antigens, and are affected by pH, temperature, and solvent.
Antibody Opsonization; Opsonization is the process by which a pathogen is marked for ingestion and destruction by a phagocyte. Opsonization involves the binding of an opsonin, e.g., antibody, to a receptor on the pathogen's cell membrane. After opsonin binds to the membrane, phagocytes are attracted to the pathogen. The Fab portion of the antibody binds to the antigen, whereas the Fc portion of the antibody binds to an Fc receptor on the phagocyte, facilitating phagocytosis. The receptor-opsonin complex can also create byproducts like C3b and C4b which are important components of the complement system. These components are deposited on the cell surface of the pathogen and aid in its destruction.
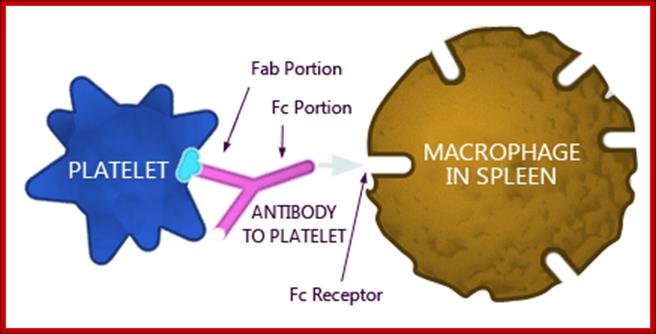
Opsonization is the process whereby an antibody attached to a virus, bacteria or cell type renders that virus, bacteria or cell type susceptible to phagocytosis. Platelets that become opsonized; https://www.slideshare.net
· Phagocytes have Fc receptors, so the opsonized cells get bound and then they are endocytosed and degraded. As part of the innate immune response, macrophages can engulf and destroy opsonized bacteria. Although this results in the clearance of bacteria, it can deprive the host of a source of antigens sufficient for the generation of an adaptive immune response (left). By virtue of the glycoprotein receptor GPIb, platelets bind deposits of C3 complement on the surface of blood-borne bacteria (right). The resulting platelet coat redirects complement-opsonized intruders from their rapid destruction by macrophages to a defined splenic DC subset, which allows priming of T cells and the establishment of immunological memory. However, these CD8α+ DCs can also serve as a beachhead for infection by facultative intracellular bacteria such as L. monocytogenes. Steffen Jung; http://www.nature.com
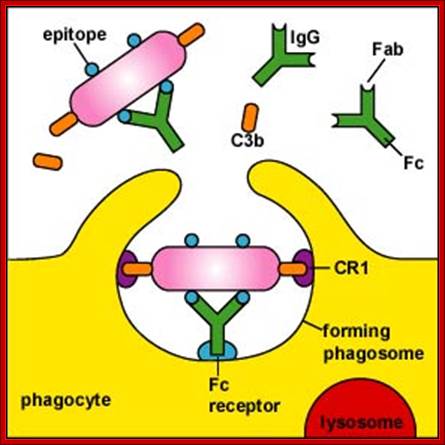
http://faculty.ccbcmd.edu
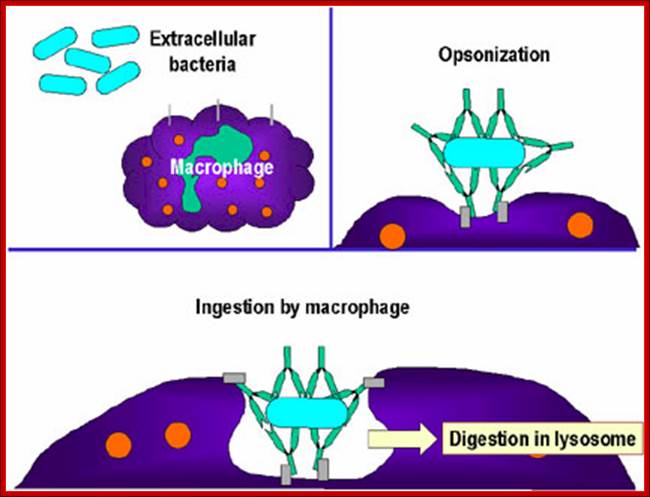
Opsonization leads to phagocytosis and then degradation the components.

An opsonin (from the Greek opsōneîn, to prepare for eating) is any molecule that enhances phagocytosis by marking an antigen for an immune response or marking dead cells for recycling (i.e., causes the phagocyte to "relish" the marked cell).[1] Opson in ancient Greece referred to the delicious side-dish of any meal, versus the sitos, or the staple of the meal.
Action of opsonins; a phagocytic cell recognizes the opsonins on the surface of an antigen and engulfs the same. Action of opsonins: a phagocytic cell recognizes the opsonin on the surface of an antigen www.digopaul.com
NK cells
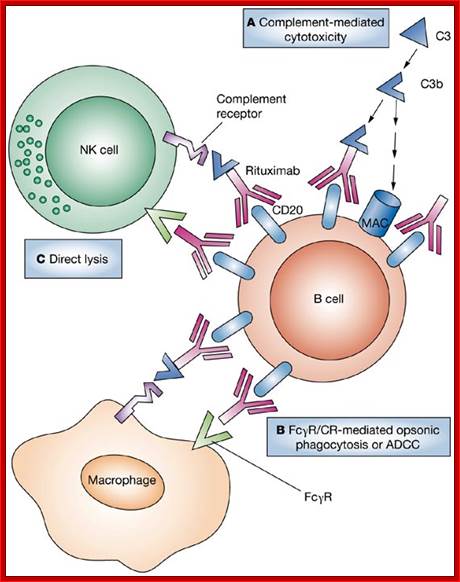
Even cells become victims to be degraded by macrophages; requires the action of NK cells
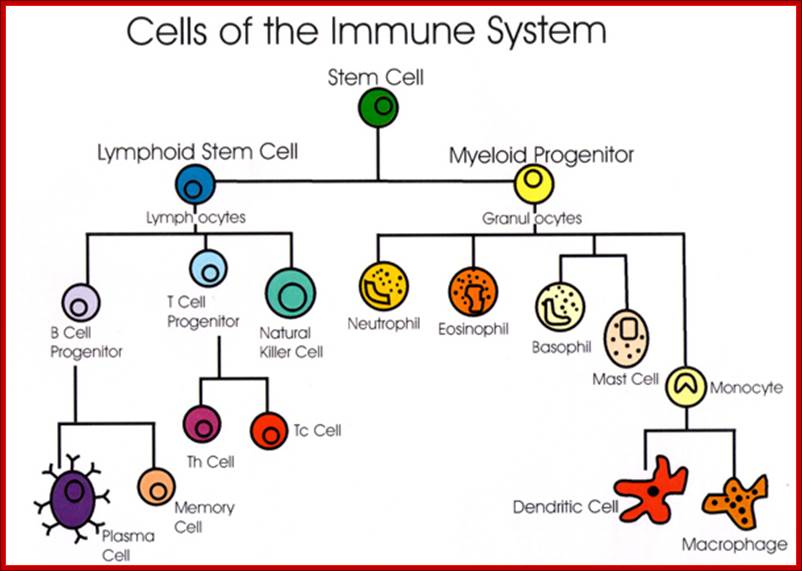
https://www.slideshare.net
Most of the immune system related cells take their origin in bone marrow as pluripotent cells. Stem cells give rise to Lymphoid Stem cells and Myeloid Progenitor cells.
Lymphoid cells differentiate into B cell progenitors, which generate B cell receptors which stick on to their membrane. These cells while circulating if they come across specific antigens or antigen carrying bacteria or viruses get activated and produce plasma cells and memory cells. Another class of Lymphocytes is T cells, which in circulation mature as Th Cells and some into T cytotoxic cells. Another set of lymphocytes develop into Natural Killer cells.
The Myeloid progenitor cells grow and develop into Granulocytes (Neutrophils, Eosinophils, Mast cells, and Monocytes differentiate into Dendrite cells and Macrophages).

Lymphocyte is a white blood cell- http://www.medfriendly.com
Granulocytes-Neutrophils, and Eosinophils,
Neutrophil, eosinophils,basophil,monocyte and lymphocyte- http://www.wisegeek.com
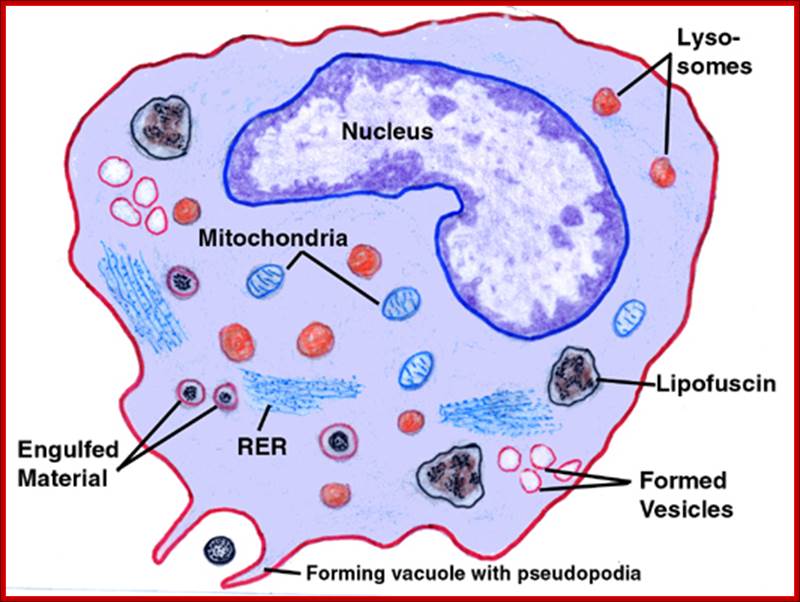
https://web.stanford.edu
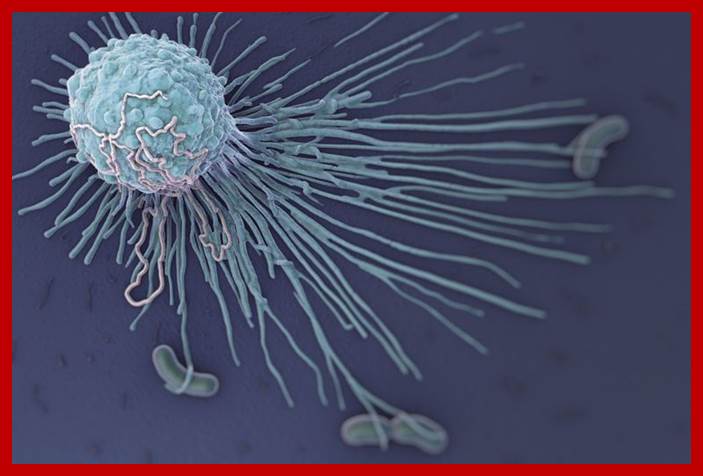
Macrophages are immune system cells that are vital to the development of non-specific defense mechanisms that provide the first line of defense against pathogens. These large immune cells are present in nearly all tissues and actively remove dead and damaged cells, bacteria, cancerous cells, and cellular debris from the body. The process by which macrophages engulf and digest cells and pathogens is called phagocytosis.Macrophages germ eating https://www.thoughtco.com
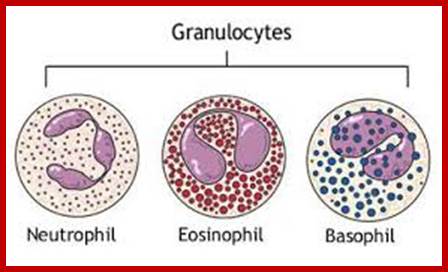
Granulocytes; http://www.sinobiological.com
Mast cells ;http://www.mastcellaware.com
Mast cells: Surface receptors and signal transduction; Michael Gurish and Mariana castells.
INTRODUCTION
Mast cells display a host of stimulatory and inhibitory surface receptors, allowing them to respond to a variety of stimuli in a modulated manner. The ultimate response of a cell to its environment is determined by the balance of stimulatory and inhibitory factors present at a given moment and acting on different receptors.
This topic review will discuss the activating and inhibitory receptors on mast cells and signal transduction mechanisms. The information in this review pertains to human mast cells whenever possible, and notation is made when data are derived purely from murine studies. Mast cell derived mediators, as well as the development, identification, and physiologic roles of mast cells are reviewed separately. (See "Mast cell derived mediators" and "Mast cells: Development, identification, and physiologic roles".)
ACTIVATING RECEPTORS
Important stimulatory receptors on the surface of mast cells include the high affinity IgE receptor, IgG receptors, toll-like receptors, and receptors for stem cell factor, complement proteins, neuropeptides and opioids.
High affinity IgE receptor — Classical mast cell activation occurs through the high affinity IgE receptor, FcEpsilonRI (FcεRI). Activation occurs when adjacent receptors, occupied by receptor-bound IgE, are cross-linked by a multivalent antigen. This is a strong stimulus for degranulation and release of preformed mediators, as well as for de novo production and subsequent release of leukotrienes, prostaglandins, and cytokines, including numerous chemokines. (See "Mast cell derived mediators".)
IgE is involved in defense against parasitic infection and in hypersensitivity reactions. It is estimated that there are 10(4)-10(6) FcεRI receptors on the surface of each mast cell [1]. As the transcript is stabilized by binding of monomeric IgE to the receptor, there is a greater number of receptors and hence capacity for binding serum IgE in atopic individuals. These likely accounts for this large range in the number of receptors per cell and the efficacy of therapy directed to reducing the serum titer of IgE [2-4]. (See 'Clinical applications' below.)
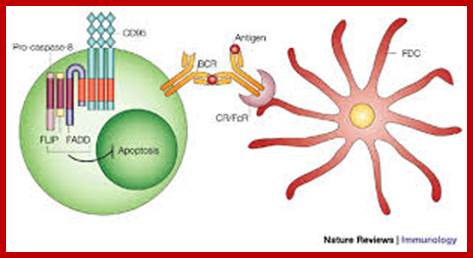
Appropriate crosslinking of the newly created
B-cell receptor (BCR) by follicular dendritic cell (FDC)-associated immune
complexes provides a mechanism to avert apoptosis by maintaining FLIP
(FAS-associated death domain (FADD) protein-like interleukin-1![]() -converting enzyme inhibitory protein) in the cytoplasm of
germinal-centre B cells. CR, complement receptor; FcR, Fc receptor Marie H. Kosco-Vilbois; http://www.nature.com
-converting enzyme inhibitory protein) in the cytoplasm of
germinal-centre B cells. CR, complement receptor; FcR, Fc receptor Marie H. Kosco-Vilbois; http://www.nature.com
Lymphocytes in the Immune System: NK, T, and B Cells, Linda Crampton;2017,
SEM of B Cells; https://hubpages.com
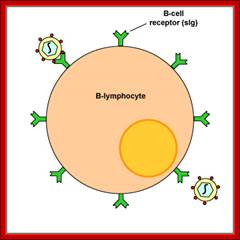
B Lymphocytes; http://www.miltenyibiotec.com
Scanning electron micrograph of a human T cell; Human T lymphocyte; https://en.wikipedia.org
T Lymphocytes (Wikipedia):
T lymphocyte pre-T cell: Lymphocytes are involved in the specific immune response and are composed mainly of precursor T cells and B cells. Pre-T cells and B cells are subsets of lymphocytes that originate in the red bone marrow from hematopoietic stem cells. Pre-T cells circulate in the blood before migrating to the thymus where they develop into specialized cells; helper T cells and killer T cells, that are able to identify antigens and infected tissue cells.
The three main
t-lymphocytes, but there are additional types (variations):
1) Helper-t-cell: This type of T lymphocyte releases Lymphokines upon
stimulation of specific antigens to promote the activation of B lymphocytes as
well as killer T lymphocytes.
2) Killer t-cell: A type of T cell that does not express markers of either T
or B-cell lineage, but may possess fc receptors for immunoglobulin g. It
functions by killing target cell through antibody-dependent cell-mediated
cytotoxicity or through perforin formation, killing cells without prior
sensitization (hence, the name).
3) Suppressor t-cell: A type of T cell that expresses CD8 transmembrane
glycoprotein, and functions by inhibiting, suppressing, or helping to stop an
immune response by releasing signals to other immune cells.
Helper T cells:
T helper cell (TH cells) assist other white blood cells in immunologic processes, including maturation of B cells into plasma cells and memory B cells, and activation of cytotoxic T cells and macrophages. These cells are also known as CD4+ T cells because they express the CD4 protein on their surface. Helper T cells become activated when they are presented with peptide antigens by MHC class II molecules, which are expressed on the surface of antigen presenting cells (APCs). Once activated, they divide rapidly and secrete small proteins called cytokines that regulate or assist in the active immune response. These cells can differentiate into one of several subtypes, including TH1, TH2, TH3, TH17, or TFH, which secrete different cytokines to facilitate a different type of immune response. Signaling from the APC directs T cells into particular subtypes.
Cytotoxic T cells:
Cytotoxic T cells (TC cells or CTLs) destroy virally infected cells and tumor cells, and are also implicated in transplant rejection. These cells are also known as CD8+ T cells since they express the CD8 glycoprotein at their surface. These cells recognize their targets by binding to antigen associated with MHC class I, which is present on the surface of nearly every cell of the body. Through IL-10, adenosine and other molecules secreted by regulatory T cells, the CD8+ cells can be inactivated to an anergic state, which prevent autoimmune diseases such as experimental autoimmune encephalomyelitis.
Memory T cells:
Memory T cells are a subset of antigen-specific T cells that persist long-term after an infection has resolved. They quickly expand to large numbers of effector T cells upon re-exposure to their cognate antigen, thus providing the immune system with "memory" against past infections. Memory T cells comprise two subtypes: central memory T cells (TCM cells) and effector memory T cells (TEM cells). Memory cells may be either CD4+ or CD8+. Memory T cells typically express the cell surface protein CD45RO.
Regulatory T cells:
Regulatory T cells (Treg cells), formerly known as suppressor T cells, are crucial for the maintenance of immunological tolerance. Their major role is to shut down T cell-mediated immunity toward the end of an immune reaction and to suppress auto-reactive T cells that escaped the process of negative selection in the thymus.
Two major classes of CD4+ Treg cells have been described — naturally occurring Treg cells and adaptive Treg cells.
Naturally occurring Treg cells (also known as CD4+CD25+FoxP3+ Treg cells) arise in the thymus and have been linked to interactions between developing T cells with both myeloid (CD11c+) and plasmacytoid (CD123+) dendritic cells that have been activated with TSLP.[4][5] Naturally occurring Treg cells can be distinguished from other T cells by the presence of an intracellular molecule called FoxP3. Mutations of the FOXP3 gene can prevent regulatory T cell development, causing the fatal autoimmune disease IPEX.
Adaptive Treg cells (also known as Tr1 cells or Th3 cells) may originate during a normal immune response.
Natural killer:
Natural killer T cells (NKT cells – not to be confused with natural killer cells) bridge the adaptive immune system with the innate immune system. Unlike conventional T cells that recognize peptide antigens presented by major histocompatibility complex (MHC) molecules, NKT cells recognize glycolipid antigen presented by a molecule called CD1d. Once activated, these cells can perform functions ascribed to both Th and Tc cells (i.e., cytokine production and release of cytolytic/cell killing molecules). They are also able to recognize and eliminate some tumor cells and cells infected with herpes viruses.
Γδ cells:
γδ T cells (gamma delta T cells) represent a small subset of T cells that possess a distinct T cell receptor (TCR) on their surface. A majority of T cells have a TCR composed of two glycoprotein chains called α- and β- TCR chains. However, in γδ T cells, the TCR is made up of one γ-chain and one δ-chain. This group of T cells is much less common (2% of total T cells) than the αβ T cells, but are found at their highest abundance in the gut mucosa, within a population of lymphocytes known as intraepithelial lymphocytes (IELs). The antigenic molecules that activate γδ T cells are still widely unknown. However, γδ T cells are not MHC restricted and seem to be able to recognize whole proteins rather than requiring peptides to be presented by MHC molecules on antigen presenting cells. Some murine γδ T cells recognize MHC class IB molecules though. Human Vγ9/Vδ2 T cells, which constitute the major γδ T cell population in peripheral blood, are unique in that they specifically and rapidly respond to a set of non-peptidic phosphorylated isoprenoid precursors, collectively named phospho-antigens. Phospho-antigens are produced by virtually all living cells. The most common phosphoantigens from animal and human cells (including cancer cells) are isopentenyl pyrophosphate (IPP) and its isomer dimethylallyl pyrophosphate (DMAPP). Many microbes produce the highly active compound Hydroxy-DMAPP (HMB-PP) and corresponding mononucleotide conjugates, in addition to IPP and DMAPP. Plant cells produce both types of phosphoantigens. Drugs activating human Vγ9/Vδ2 T cells comprise synthetic phosphoantigens and amino bisphosphonates, which up-regulate endogenous IPP/DMAPP
T cell development in the thymus:
All T cells originate from haematopoietic stem cells in the bone marrow. Haematopoietic progenitors derived from haematopoietic stem cells populate the thymus and expand by cell division to generate a large population of immature thymocytes. The earliest thymocytes express neither CD4 nor CD8, and are therefore classed as double-negative (CD4-CD8-) cells. As they progress through their development they become double-positive thymocytes (CD4+CD8+), and finally mature to single-positive (CD4+CD8- or CD4-CD8+) thymocytes that are then released from the thymus to peripheral tissues.
About 98% of thymocytes die during the development processes in the thymus by failing either positive selection or negative selection, whereas the other 2% survive and leave the thymus to become mature immuno-competent T cells.
The thymus contributes fewer cells as a person ages. As the thymus shrinks by about 3% a year throughout middle age, there is a corresponding fall in the thymic production of naive T cells, leaving peripheral T cell expansion to play a greater role in protecting older subjects.
Positive selection:
Positive selection "selects for" T cells capable of interacting with MHC. Double-positive thymocytes (CD4+/CD8+) move deep into the thymic cortex where they are presented with self-antigens. These self-antigens are expressed by thymic cortical epithelial cells that express Activation-Induced (Cytidine) Deaminase on both MHC molecules on the surface of cortical cells. Only those thymocytes that interact with MHC-I or MHC-II will receive a vital "survival signal." All that can't will die by apoptosis. This process insures that the TCR on affinity cannot serve useful functions in the body (i.e. the cells must be able to interact with MHC and peptide complexes in order to effect immune responses). The vast majority of all thymocytes end up dying during this process.
A thymocyte's fate is also determined during positive selection. Double-positive cells (CD4+/CD8+) that are positively selected on MHC class II molecules will eventually become CD4+ cells, while cells positively selected on MHC class I molecules mature into CD8+ cells. A T cell becomes a CD4+ cell by down regulating expression of its CD8 cell surface receptors. If the cell does not lose its signal through the ITAM pathway, it will continue down-regulating CD8 and become a CD4+, single positive cell. But if there is a signal interruption, the cell stops downregulating CD8 and switches over to downregulating CD4 molecules instead, eventually becoming a CD8+, single positive cell.
This process does not remove thymocytes that may cause autoimmunity. The potentially autoimmune cells are removed by the process of negative selection (discussed below).
Negative selection:
Negative selection removes thymocytes that are capable of strongly binding with "self" peptides presented by MHC. Thymocytes that survive positive selection migrate towards the boundary of the thymic cortex and thymic medulla. While in the medulla, they are again presented with self-antigen in complex with MHC molecules on thymic epithelial cells.[8] Thymocytes that interact too strongly with the antigen receive an apoptotic signal that leads to cell death. However, some of these cells are selected to become Treg cells. The remaining cells exit the thymus as mature naive T cells. This process is an important component of central tolerance and serves to prevent the formation of self-reactive T cells that are capable of inducing autoimmune diseases in the host.
In summary, β-selection is the first checkpoint, where the T cells which are able to form a functional TCRβ chain and bind to pre-Tα are allowed to proceed. Next, positive selection checks that T cells have successfully re-arranged their TCRα locus and are capable of recognizing peptide-MHC complexes. Negative selection then removes for T cells that bind too strongly to self-antigens. These selection processes allow for tolerance of self by the immune system.
Activation; NCBI.NIH.NLM.gov
Helper T Cells and Lymphocyte Activation
Helper T cells are arguably the most important cells in adaptive immunity, as they are required for almost all adaptive immune responses. They not only help activate B cells to secrete antibodies and macrophages to destroy ingested microbes, but they also help activate cytotoxic T cells to kill infected target cells. As dramatically demonstrated in AIDS patients, without helper T cells we cannot defend ourselves even against many microbes that are normally harmless.
Helper T cells themselves, however, can only function when activated to become effector cells. They are activated on the surface of antigen-presenting cells, which mature during the innate immune responses triggered by an infection. The innate responses also dictate what kind of effector cell a helper T cell will develop into and thereby determine the nature of the adaptive immune response elicited.
In this final section, we discuss the multiple signals that help activate a T cell and how a helper T cell, once activated to become an effector cell, helps activate other cells. We also consider how innate immune responses determine the nature of adaptive responses by stimulating helper T cells to differentiate into either TH1 or TH2 effector cells.
To activate a cytotoxic or helper T cell to proliferate and differentiate into an effector cell, an antigen-presenting cell provides two kinds of signals.
Signal 1 is provided by a foreign peptide bound to an MHC protein on the surface of the presenting cell. This peptide-MHC complex signals through the T cell receptor and its associated proteins. Signal 2 is provided by costimulatory proteins, especially the B7 proteins (CD80 and CD86), which are recognized by the co-receptor protein CD28 on the surface of the T cell. The expression of B7 proteins on an antigen-presenting cell is induced by pathogens during the innate response to an infection. Effector T cells act back to promote the expression of B7 proteins on antigen-presenting cells, creating a positive feedback loop that amplifies the T cell response.
Signal 2 is thought to amplify the intracellular signaling process triggered by signal 1. If a T cell receives signal 1 without signal 2, it may undergo apoptosis or become altered so that it can no longer be activated, even if it later receives both). This is one mechanism by which a T cell can become tolerant to self-antigens.
The two signals that activate a helper T cell: (A) A mature antigen-presenting cell can deliver both signal 1 and 2 and thereby activate the T cell. (B) An immature antigen-presenting cell delivers signal 1 without signal 2, which can kill or inactivate.
The T cell receptor does not act on its own to transmit signal 1 into the cell. It is associated with a complex of invariant transmembrane proteins called CD3, which transduces the binding of the peptide-MHC complex into intracellular. In addition, the CD4 and CD8 co-receptors play important parts in the signaling process, as illustrated in Figure 24-64.
The T cell receptor and its associated CD3 complex: All of the CD3 polypeptide chains (shown in green), except for the ζ (zeta) chains, have extracellular Ig-like domains and are therefore members of the Ig superfamily.
The signaling events initiated by the binding of peptide-MHC complexes to T cell receptors (signal 1) when T cell receptors are clustered by binding to peptide-MHC complexes on an antigen-presenting cell, CD4 molecules on helper cells or CD8 molecules.
The combined actions of signal 1 and signal 2 stimulate the T cell to proliferate and begin to differentiate into an effector cell by a curiously indirect mechanism. In culture, they cause the T cells to stimulate their own proliferation and differentiation by inducing the cells to secrete a cytokine called interleukin-2 (IL-2) and simultaneously to synthesize high affinity cell-surface receptors that bind it. The binding of IL-2 to the IL-2 receptors activates intracellular signaling pathways that turn on genes that help the T cells to proliferate and differentiate into effector cells. As discussed in Chapter 15, there are advantages to such an autocrine mechanism. It helps ensure that T cells differentiate into effector cells only when substantial numbers of them respond to antigen simultaneously in the same location, such as in a lymph node during an infection. Only then do IL-2 levels raise high enough to be effective.
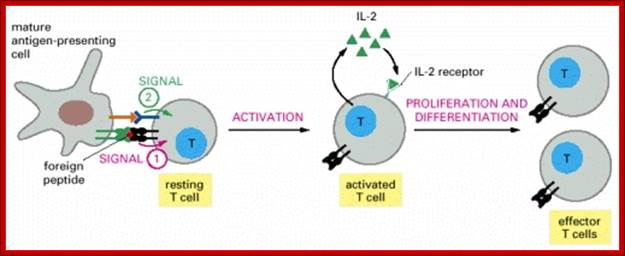
https://www.slideshare.ne
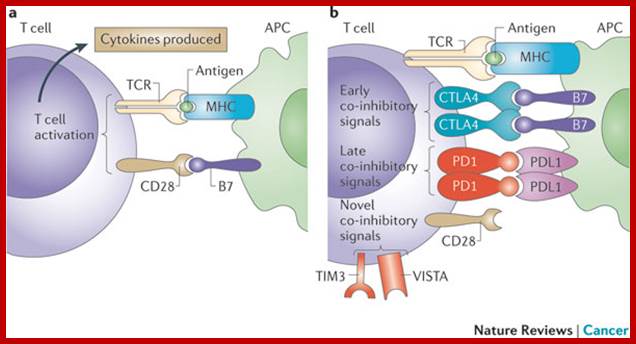
Basic mechanisms of T cell stimulation and inhibition. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps; a | T cell activation begins with interaction of the T cell receptor (TCR) on a T cell with major histocompatibility complex (MHC) bound to antigen on an antigen-presenting cell (APC). This is known as signal 1, but appropriate activation of the T cell requires additional signals that are provided by the interaction between CD28 and B7 (signal 2). b | T cell activation is limited by cytotoxic T lymphocyte-associated protein 4 (CTLA4), which is upregulated on activated T cells, where it outcompetes CD28 for binding to B7 on an APC. Additional regulation of T cell activity is also provided by later inhibitory signals through other molecules such as programmed cell death 1 (PD1), which binds to PD1 ligand 1 (PDL1). Other regulators of T cell activation have recently been characterized and may have important roles; these regulators include T cell immunoglobulin and mucin domain-containing protein 3 (TIM3; also known as HAVCR2) and V-domain immunoglobulin suppressor of T cell activation (VISTA) Padmanee Sharma, Klaus Wagner, Jedd D. Wolchok & James P. Allison; http://www.nature.com
Activation of CD4+ T cells occurs through the engagement of formation of the T cell receptor and CD28 on the T cell by the major histocompatibility complex (MHC) peptide and B7 family members on the APC, respectively. Both are required for production of an effective immune response; in the absence of CD28 co-stimulation, T-cell receptor signaling alone results in anergy. The signaling pathways downstream from both CD28 and the T cell receptor involve many proteins.
The first signal is provided by binding of the T cell receptor to a short peptide presented by the major histocompatibility complex (MHC) on another cell. This ensures that only a T cell with a TCR specific to that peptide is activated. The partner cell is usually a professional antigen presenting cell (APC), usually a dendritic cell in the case of naïve responses, although B cells and macrophages can be important APCs. The peptides presented to CD8+ T cells by MHC class I molecules are 8–9 amino acids in length; the peptides presented to CD4+ cells by MHC class II molecules are longer, usually 12–25 amino acids in length,[9] as the ends of the binding cleft of the MHC class II molecule are open.
The second signal comes from co-stimulation, in which surface receptors on the APC are induced by a relatively small number of stimuli, usually products of pathogens, but sometimes breakdown products of cells, such as necrotic-bodies or heat shock proteins. The only co-stimulatory receptor expressed constitutively by naïve T cells is CD28, so co-stimulation for these cells comes from the CD80 and CD86 proteins, which together constitute the B7 protein, (B7.1 and B7.2 respectively) on the APC. Other receptors are expressed upon activation of the T cell, such as OX40 and ICOS, but these largely depend upon CD28 for their expression. The second signal licenses the T cell to respond to an antigen. Without it, the T cell becomes anergic, and it becomes more difficult for it to activate in future. This mechanism prevents inappropriate responses to self, as self-peptides will not usually be presented with suitable co-stimulation.
The T cell receptor exists as a complex of several proteins. The actual T cell receptor is composed of two separate peptide chains, which are produced from the independent T cell receptor alpha and beta (TCRα and TCRβ) genes. The other proteins in the complex are the CD3 proteins: CD3εγ and CD3εδ heterodimers and, most important, a CD3ζ homodimer, which has a total of six ITAM motifs. The ITAM motifs on the CD3ζ can be phosphorylated by Lck and in turn recruit ZAP-70. Lck and/or ZAP-70 can also phosphorylate the tyrosine on many other molecules, not least CD28, LAT and SLP-76, which allows the aggregation of signaling complexes around these proteins.
Phosphorylated LAT recruits SLP-76 to the membrane, where it can then bring in PLC-γ, VAV1, Itk and potentially PI3K. Both PLC-γ and PI3K act on PI(4,5)P2 on the inner leaflet of the membrane to create the active intermediaries diacylglycerol (DAG), inositol-1,4,5-trisphosphate (IP3), and phosphatidlyinositol-3,4,5-trisphosphate (PIP3). DAG binds and activates some PKCs, most important, in T cells PKCθ, a process important for activating the transcription factors NF-κB and AP-1. IP3 is released from the membrane by PLC-γ and diffuses rapidly to activate receptors on the ER, which induce the release of calcium. The released calcium then activates calcineurin, and calcineurin in turn activates NFAT, which then translocate to the nucleus. NFAT is a transcription factor, which activates the transcription of a pleiotropic set of genes, most notable, IL-2, a cytokine that promotes long term proliferation of activated T cells.
PLCγ can also initiate the NF-κB pathway. DAG activates PKCθ, which then phosphorylates CARMA1 causing it to unfold and function as a scaffold. The cytosolic domains bind an adapter Cbl 10 via CARD (Caspase activation and recruitment domains) domains; that then binds TRAF6, which is ubiquitinated at K63.
Template:Rp:513–523 This form of ubiquitination does not lead to degradation of target proteins. Rather it serves to recruit NEMO, IKKα and β, and TAB1-2/ TAK1. TAK 1 phosphorylates IKK-β, which then phosphorylates IκB allowing for K48 ubiquitination: leads to proteasomal degradation. Rel A and p50 can then enter the nucleus and bind the NF-κB response element. This coupled with NFAT signaling allows for complete activation of the IL-2 gene.
While in most cases activation is dependent on TCR recognition of antigen, alternative pathways for activation have been described. For example, cytotoxic T cells have been shown to become activated when targeted by other CD8 T cells leading to tolerization of the latter.
|
Cytokine or Chemokine |
Producing cell |
Target cell |
Effect |
|
Interleukin-1 (IL-1) |
Macrophages, monocytes and B cells |
T cells, B cells |
Acts as a growth regulator of T cells and B cells. Induces other cells such as hepatocytes to produce proteins relevant to host defense Serves as an endogenous pyrogen, which produces fever. |
|
Interleukin-2 (IL-2) |
T cells |
T cells |
Stimulates the proliferation of T cells and activates natural killer cells. |
|
Interleukin-3 (IL-3) |
Stem cells, Mast cells |
Regulates the proliferation of stem cells and the differentiation of mast cells. |
|
|
Interleukin-4 (IL-4) |
TH2 |
B cells |
B cell proliferation and enhanced antibody synthesis. |
|
Interleukin-5 (IL-5) |
TH2 |
B cells |
B cell differentiation and IgA synthesis |
|
Interleukin-6 (IL-6) |
TH2, monocytes, macrophages |
B cells, plasma cells, stem cells |
B cell differentiation and antibody production. T cell activation, growth and differentiation. Has a major role in the mediation of the inflammatory and immune responses initiated by infection or injury. |
|
CXCL-8 |
Many host cells |
T cells, neutrophils, macrophages |
Chemo attractant for neutrophils |
|
α-Interferon (IFN-α) |
Leucocytes, tissue cells |
Tissue cells |
Inhibition of viruses |
|
γ-Interferon (IFN-γ) |
T cells |
Tissue cells, macrophages, natural killer cells |
Inhibition of protein synthesis in virally infected cells. Activation of macrophages and natural killer cells. Stimulates IL-1, IL-2 and antibody production. |
|
Tumor Necrosis Factor-α (TNF-α) |
T cells |
Tissue cells (tumors) |
Kills cells, including tumor cells |
|
Tumor Necrosis Factor-β (TNF-β) |
T cells |
Tissue cells (tumors) |
Kills cells, including tumor cells |
|
Colony Stimulating Factors (CSF) |
TH1, macrophages |
Phagocytes |
Causes phagocytic white cells of all types to differentiate and divide. |
|
Macrophage chemo attractant and activating factor (MCAF) |
Monocytes, macrophages, fibroblasts (connective tissue cells) and keratinocytes (skin cells) |
Macrophages, T cells |
Attract and activate macrophages and T cells. |
|
B cell growth factors |
T cells |
B cells |
Multiplication of B cells |
Important cytokines and chemokines in the immune response; A large number of signal molecules are produced by various tissues in the body. The cytokines and chemokines listed here are important in various parts of the immune response.
Example of Cytokines and Lymphokines action: Natural killer cell activation involves interferon or IL-2, and IgG
- Natural killer cells are activated when a chemokine (either interferon or IL-2) is present and IgG is recognized on the surface of a cell.
- This causes the natural killer cell to attack, forming a pore in the target cell and pumping lysosomal contents into it causing its death.
We now again return to natural killer cells. Natural killer cells cannot be induced as cytotoxic T cells are and their activation comes about in a different manner. The mechanism of natural killer attack involves several separate activation steps. Interferon from virally infected cells or IL-2 from T cells must first activate natural killer cells. The second activation step occurs when the natural killer cells encounters a target cell coated with IgG and docks with its Fc region. Having several activation steps prevents the natural killer cell from mistakenly attacking normal cells. Docking then causes changes in the location of cytoplasmic granules and Golgi apparatus within the natural killer cell to a location close to the edge of the cytoplasmic membrane and near the target cell. At this point, the natural killer cell is activated and ready to attack its target. A pore-forming protein, perforin 1 is inserted into the membrane of the target and the natural killer cell then exports lysosomal secretions into the target cell. The pore made in the membrane together with the attack of the lysosomal contents eventually causes the target cell to lyse and die. The natural killer cell then prepares its cytoplasmic components for the next victim.
After successful docking, the helper T cell releases a class of chemical messenger called cytokines. This achieves the following: it causes the helper T cells to multiply, and stimulates both the APC and the helper T cell to exchange chemical messages between themselves and with other cells of the immune system.
The helper T cell also releases a cytokine called interleukin-2 (IL-2): this cytokine has panoply of immune functions, one of which is the proliferation of lymphocytes following activation by a specific antigen. The APC releases cytokines called interleukin-1 (IL-1) and tumor necrosis factor (TNF). The latter steps up the production of IL-1 and perform many of the same functions as IL-1, including the induction of fever in Brian so that his body can assist in fighting off the bacterial infection more effectively.
The proliferating helper T cells release substances that signal another type of lymphocyte, the B cell (that also specifically recognizes the antigen), to begin multiplying and differentiating into antibody-producing cells. This initiates the effector phase of the humoral immune response.
The antibodies released by the B cells bind in a lock-and-key fashion to antigens on the surfaces of bacterial invaders that survived the initial attack by macrophages and bacterial products. The binding of the antibody to the bacterial antigens achieve two things first, it makes it easier for “killer” cells to attack and destroy the invading bacteria by both phagocytosis and the release of other factors that can directly lyse the bacteria. Second, it activates another immune military unit called complement—a group of proteins that act like the special forces of the immune defense system because their duty is to begin the lethal process of punching holes in the cell walls of the residual invading bacterial army. The humoral immune response is summarized in the figure below.
Macrophages and Dendritic cells are phagocytes and are also responsible for "presenting" antigens to T cells to initiate both cell-mediated and antibody-mediated adaptive immune responses. The interaction of PAMPs and TLRs on dendritic cells causes them to secrete cytokines, including
- Interleukin 12 (IL-12) which stimulates the production of Th1 cells;
- Interleukin 23 (IL-23) which stimulates the production of Th17 cells;
- Interleukin 6 (IL-6), which interferes with the ability of regulatory T cells to suppress the responses of effector T cells to antigen. A double-
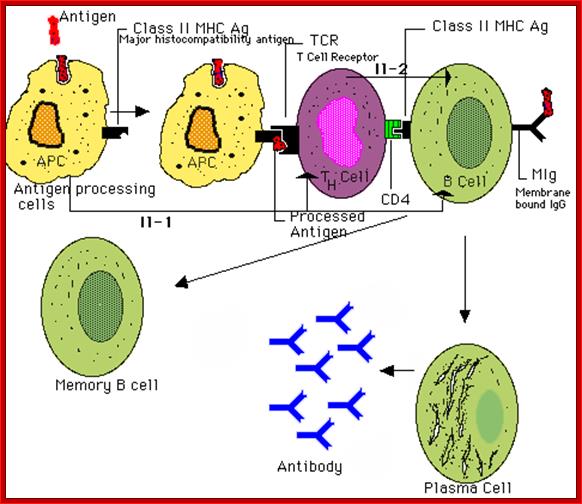
After successful docking, the helper T cell releases a class of chemical messenger called cytokines. This achieves the following: it causes the helper T cells to multiply, and stimulates both the APC and the helper T cell to exchange chemical messages between themselves and with other cells of the immune system.
The B cells thus activated undergo proliferation to produces a specific Igs against the activated antigen. At the same time they also generate memory cells, which can remain for a long time to be activated immediately on facing that antigen.
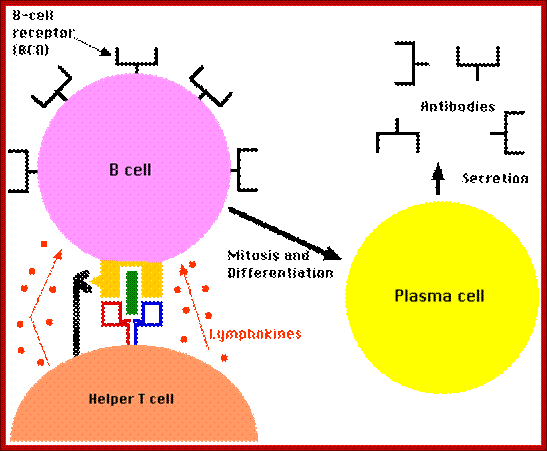
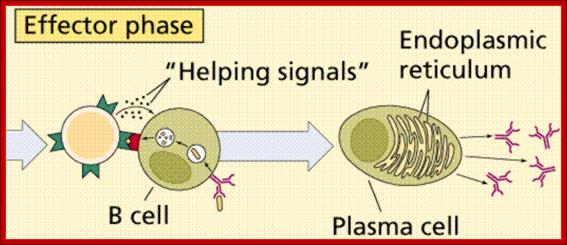
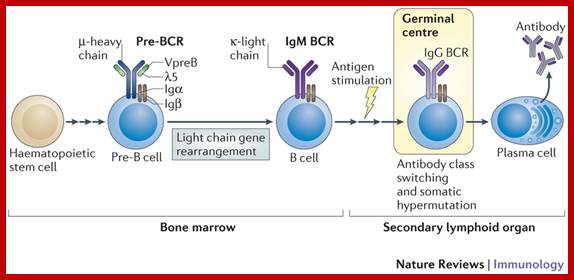
Max D. Cooper; ;http://www.nature.com
The activation phase- It begins when invading bacteria are phagocytized by an antigen-presenting cell (APC), such as a dendritic cell or macrophage. The bacterium is digested and its antigens processed and presented in combination with the MHC Class II complex on the surface of the APC.
The antigen-MHC complex is recognized by a type of immune cell called a CD4+ or helper T cell (Th). The helper T cell begins the attack by docking its antigen receptor to the displayed antigen. The docking process requires the presence of co-stimulatory molecules like B7 and CD28.
APC cells
|
Macrophage |
Dendritic Cell |
B cell |
|
|
MHC-II Expression |
Low levels. |
Always Expressed. |
Always Expressed. |
|
Antigen type and |
Extracellular Antigens: |
Intracellular & |
Extracellular Antigen binds |
|
Co-Stimulation |
Low levels. |
Always expressed |
Low levels. |
|
Location |
Lymphoid tissue |
Lymphoid tissue |
Lymphoid tissues. |
Opsonization is the process whereby an antibody attached to a virus, bacteria or cell type renders that virus, bacteria or cell type susceptible to phagocytosis. Platelets that become opsonized by IgG antibodies in ITP will undergo phagocytosis and will be destroyed.
The spleen and the reticuloendothelial system (RES) are the components of the immune system responsible for removal of opsonized platelets
Cellular Immunity:
Cell-mediated immunity (Wikipedia) is an immune response that does not involve antibodies but rather involves the activation of phagocytes, natural killer cells (NK), antigen-specific cytotoxic T-lymphocytes, and the release of various cytokines in response to an antigen. Historically, the immune system was separated into two branches: humoral immunity, for which the protective function of immunization could be found in the humor (cell-free bodily fluid or serum) and cellular immunity, for which the protective function of immunization was associated with cells. CD4 cells or helper T cells provide protection against different pathogens. Cytotoxic T cells cause death by apoptosis without using cytokines; therefore in cell mediated immunity cytokines are not always present. (Remember, B cells and their products make up the first branch: humoral immunity.) Learning about T cells tends to feel less straightforward than learning about B cells—it can be a frustrating alphabet soup of terms, (CD, MHC, HLA…) so before we get started, it is worth taking some time to define the different types of T cells and the receptors on each.
The CD nomenclature: T lymphocytes come in two flavors: helper and cytotoxic. Although there are a few smaller populations of other flavors of T cells, (regulatory T cells, natural killer T cells…etc which are the subject of current research) for now, let’s just focus on the two: helper T cells and cytotoxic T cells. Helper T cells are so-named because their main function is to secrete chemical mediators that direct, or “help” other T cells, B cells and/or phagocytes to mount a potent response against microbes. Cytotoxic T cells are so-named because when they become activated against infected cells, they directly kill those cells. When immunologists began to study different kinds of lymphocytes, they began to classify them by the markers that were on their surfaces. As it turns out, helper T cells express a protein in the “CD” family called “CD4” on their surfaces while cytotoxic T cells express a different but related protein called “CD8” on their surfaces. Thus, CD4+ T cells are helper T cells; CD8+ T cells are cytotoxic T cells. You will see helper and cytotoxic T cells described in both ways, but you should know that these terms are synonymous. The illustration to the left shows helper and cytotoxic T cells with their associated CD proteins. In this module, however, helper and cytotoxic T cells will be represented without these CD proteins as orange and red lymphocytes, respectively.
Major Histocompatibility Complex: Unlike B cells, which recognize circulating antigens of many chemical structures, the vast majority of T cells (>95%) are only able to recognize peptide fragments that are displayed by specialized molecules called MHC molecules on the surfaces of antigen presenting cells. More alphabet soup. MHC stands for major histocompatibility complex and, much like T cells, MHC molecules come in two flavors: MHC I and MHC II. [In reading about immunology, you may come across the abbreviation HLA used in the same context. Don’t worry, HLA describes the same genetic locus and molecules as MHC, but is specific to humans; MHC is a more general term that applies to all species.] Understanding the differences between MHC I and MHC II is critical. This is described in the following two paragraphs and summarized in a table.
MHC I: MHC I molecules are found on all healthy nucleated cells in the body. MHC I molecules are designed to display peptide antigens that are found within the cytoplasm of cells. So, for example, if a virus invaded a cell in your liver, the MHC I molecules on the surface of that liver cell would display peptide pieces of that virus on the cell surface. If a virus invaded the cytoplasm of a white blood cell, that cell would display peptide antigens of the virus on its surface. MHC I molecules and the antigens they present are mainly recognized by CD8+ cytotoxic T cells (and not by CD4+ cells). Thus, CD8+ cytotoxic T cells are better than CD4+ helper T cells at responding to viruses and microbes that can escape from vesicles into the cytosol because the peptide antigens of these microbes are displayed on MHC I molecules.
MHC II: In contrast, MHC II molecules are not found everywhere. MHC II molecules are restricted to specialized types of cells, namely antigen presenting cells such as dendritic cells, macrophages and B cells. Unlike MHC I molecules, which display antigens from the cytosol, MHC II molecules display antigens from microbes within the cell’s vesicles. So for instance, let’s say you got a cut on your finger and some bacteria got inside. A dendritic cell at the site of the cut would phagocytose the bacteria and peptide pieces of the bacteria would be displayed on the dendritic cell via MHC II molecules. MHC II molecules and the antigens they display are mainly recognized by CD4+ helper T cells (not by CD8+ cells).
The image below is meant to help summarize and review the properties of MHC I and MHC II molecules. Click on the row or column titles in the image to fill in their respective fields. These are key to understanding the "bigger picture" in cell-mediated immunity--see if you can do it from memory.
Cellular immunity protects the body by:
- Activating antigen-specific cytotoxic T-lymphocytes that are able to induce apoptosis in body cells displaying epitopes of foreign antigen on their surface, such as virus-infected cells, cells with intracellular bacteria, and cancer cells displaying tumor antigens;
- Activating macrophages and natural killer cells, enabling them to destroy pathogens; and
- Stimulating cells to secrete a variety of cytokines that influence the function of other cells involved in adaptive immune responses and innate immune responses.
Naïve T lymphocytes recognize antigens and they are activated in peripheral lymph system. Like the humoral immune response, this is also divided into activation and effector phases. This leads to the production of antigen specific cell pools. These become effector and memory T cells.
Virus infected several epithelial cell types is processed and presented as MHCI class proteins. A helper T cell with CD$ binds MHCII antigen complex which is recognized my T CD4 cells. The activated Th cell releases few cytokines like IL2 and Interferon gamma IFN-g.
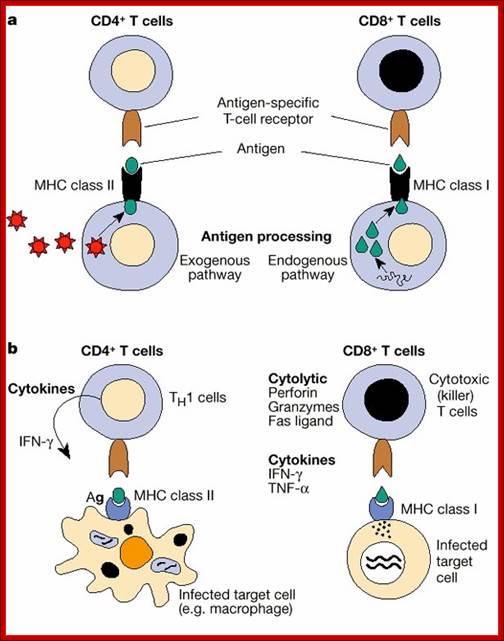
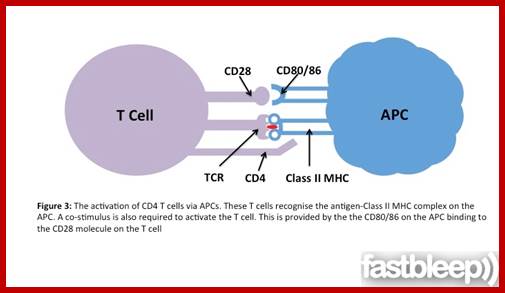
Different class of T cells bind to APCs with different MHC class proteins; https://www.fastbleep.com/
Cell mediated immunity by Effector mechanism:
CM1 is adaptive immune response against intracellular microbes. It is mediated by T cells and can be transferred from immunized to naïve individuals to T cells and not by antibodies. The effector phase starts when activated Cytotoxic T cells (CD8) which stimulate proliferation by cytokine IL-2, which recognize MHCI type. The cytokines also attract Killer T cells to the site of infection and binds to MHC I proteins on epithelia (infected) and they release perforins which perforate cells to lyse and die. Once the effector cells bring about the killing of infected cells, the regulatory T cells turn off activated cytotoxic T-cell. The memory Tcell responds quickly to the second viral attack. Those immune cells remain active, after the death most of the T cells with time, are called memory cells. This explanation is not restricted to virus, this applies to other pathogens.
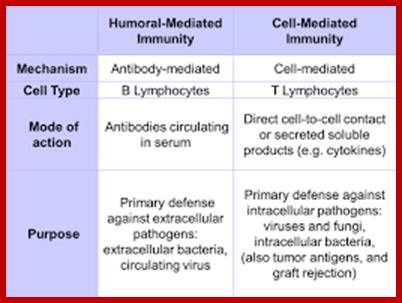
http://weishendopublications.com
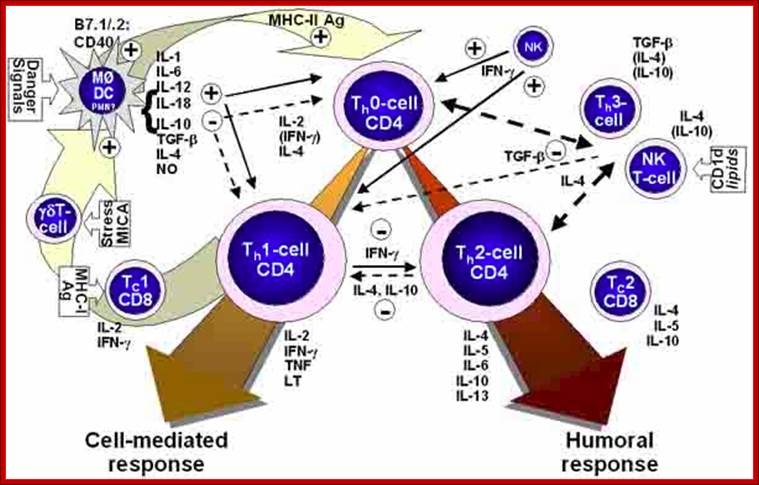
Figure. Postulated inter-relationship of T-helper-cells (Th) (Th0-, Th1-, Th2-, Th3-cells), T-regulatory-cells (T-reg), gamma-delta-T-cells, natural-killer-T- (NKT) cell or cytotoxic-T-cell (Tc) -1 and -2 and the cytokines they express in the regulation/development of a competent cell-mediated immune response. See text for detailed description. https://www.slideshare.net
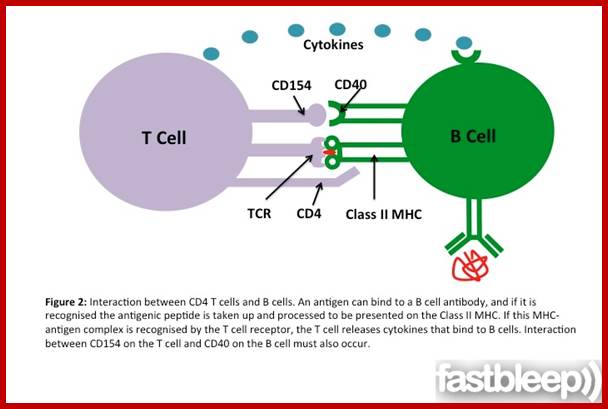
www.fastbleep.com
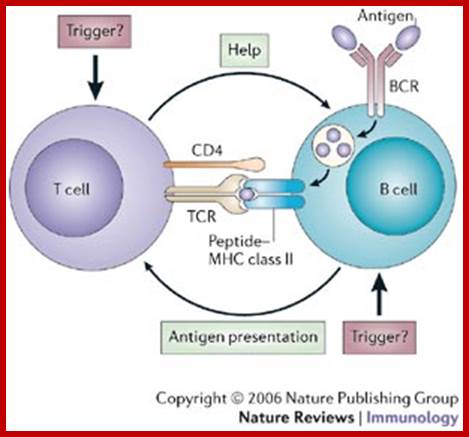
B-cell–T-cell interactions. Jonathan C. W. Edwards & Geraldine Cambridge; The two-way interaction between B cells and T cells provides the basis for the concept that, in certain autoimmune diseases, an amplification cycle might allow persistent immunopathology to arise from a minor 'trigger' factor. Such a trigger might initiate the cycle through events in either the B-cell or the T-cell compartment, including the stochastic generation of both B-cell receptors (BCRs) and T-cell receptors TCRs).; http://www.nature.com/
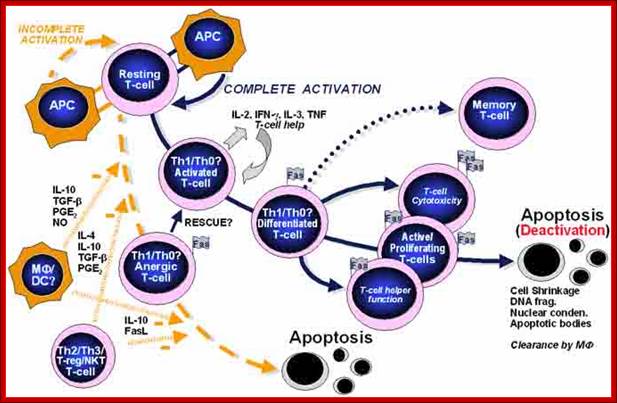
Figure. Postulated steps (activation, anti-inflammatory induced suppression, maladaptive intracellular signaling, pathologic induction of apoptosis) at which the process of immune cell (T-cell) activation/differentiation might be inhibited/suppressed by various agents leading to a resolution/suppression of the response to foreign antigen in T-lymphocytes following the onset of sepsis. https://www.slideshare.net
There are two types (main) cell mediated immunity. In one, which is exemplified by DTH reactions, CD4+ Th1 Cells, as well as CD8 Th2 cells, recognize antigens of microbes that have been ingested by phagocyte and activate the phagocytes to kill the microbes. In the second type of CMI, CD8+ CTLs kill any nucleated cell that contains foreign antigens (microbial /tumor antigens) in cytosol.
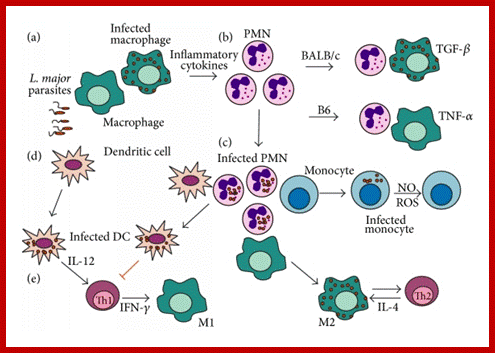
www.researchgate.net
Phagocytes act as effector system in killing microbes:
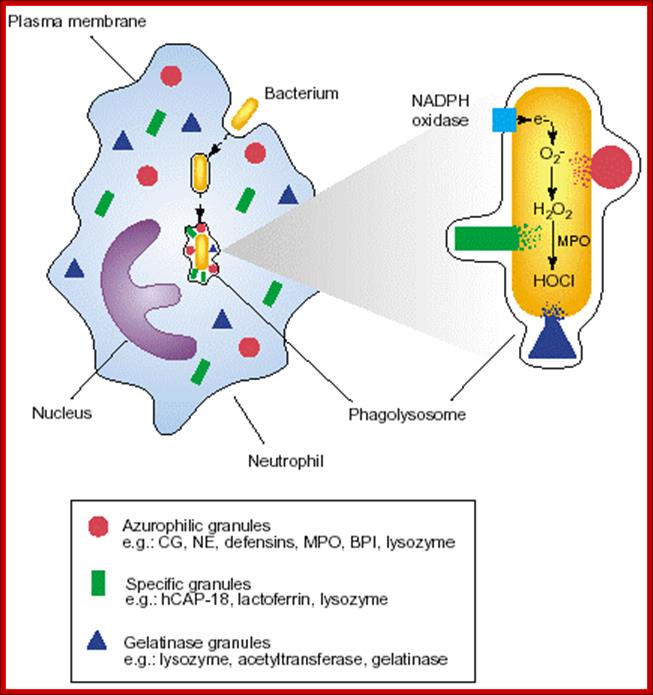
Effector NFkB activation and its effect; https://www.life.illinois.edu
Costimulation
The binding of a T cell to an antigen-presenting cell (APC) is by itself not enough to activate the T cell and turn it into an effector cell: one able to, for examples,
- kill the APC (CD8+ cytotoxic T lymphocytes [CTLs])
- carry out cell-mediated immune reactions (CD4+ Th1 cells)
- provide help to B cells (CD4+ Th2 cells)
In order to become activated, the T cell must not only bind to the epitope (MHC-peptide) with its TCR but also receive a second signal from the APC. The receipt of this second signal is called Costimulation. Among the most important of these costimulators are molecules on the APC designated B7 and their ligand on the T cell designated CD28. The binding of CD28 to B7 provides the second signal needed to activate the T cell.
Although T cells may encounter self-antigens in body tissues, they will not respond unless they receive a second signal. In fact, binding of their TCR ("signal one") without "signal two" causes them to self-destruct by apoptosis. Most of the time, the cells presenting the body's own antigens either
- fail to provide signal two
- transmit an as-yet-unidentified second signal that turns the T cell into a regulatory T cell (Treg) that suppresses immune responses.
Cell mediated immune response consists of several steps; naive cell recognition of cell- Associated antigens in peripheral lymphoid organs, clonal expansion of T cells and their differentiation into effector cells.
Migration of affecter cells to the site of infection or antigen challenge and elimination of microbes or antigen.
![]()
Kimbels biology pages
CD4 helper T lymphocytes may differentiate into specialized effector cells Th1 cells that secrete IFN-g which favors phagocyte mediated immunity, or into Th2 cells that secrete Il-1and Il4 and Il5, which favor IgE and eosinophil/ mast cell mediated immune reaction. The differentiation of naïve CD4+Tcells into Th1 and Th2 population is controlled by cytokines produced by the T cells themselves.
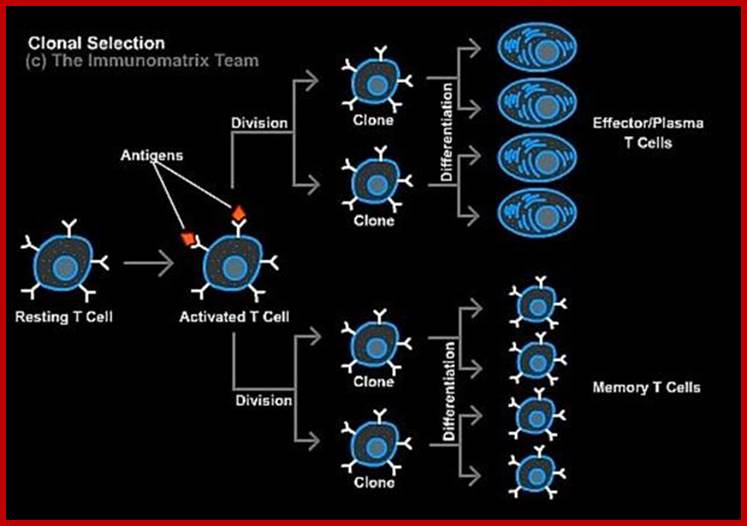
CD8+ T cells differentiate in to effector CTLs Acquiring the capacity to kill targets, under the influence of co stimulators and help from CD4+T c ells. www.dianliwenmi.com/postimg_4179016.html
The migration of T cells to sites of infection is mediated by chymykines and the binding of adhesion molecules to their ligands on activated endothelium.
The activation of macrophages by TH1 cells is mediated by IFN-g and CD40L-CD40 interactions. Activated macrophages kill phogocytosed microbes, stimulate inflammation, and repair damaged tissues. If the infection is not fully resolved activated macrophages cause tissue damage and fibrosis.
CD8+CTLs kill cells that express peptides derived from cytosolic antigens, e.g. viral antigens that are presented in association with class I MHC molecules. CTL – mediated killing is mediated mainly by granule exocytosis, which release granzymes and perforin. Perforin facilitates granzymes entry into the cytoplasm of target cells and granzymes initiate several pathways of apoptosis. CTLs also express FasL, which engages Fas on target cell membranes and trigger apoptosis of target cells.