Regulation of Gene Expression:
Introduction:
General Features of Regulation:
Molecular Genetics:
The term encompasses molecular based genetics; it is the ultimate form of information that we can get from chromosomes; chromosomal located genes to down-stream product such as mRNA, nc RNAs to proteins. Most of the ncRNAs, as they are coded for by the DNA, are involved in protein, RNA and gene regulation; there is lot to be learnt about them.
Analysis of molecular Genetics is characterized by different terminologies which provide related information; this information is called OMICs.
Flow of information:
Chromosomes (genome)-> mRNAs> proteins> metabolism> phenotype.
Chromosomes (genome)-> ncRNAs > protein synthesis >metabolism> phenotype.
Chromosomes (genome)-> ncRNAs > regulatory.
Chromosomes (genome)-> mRNA + ncRNA (functional) -> metabolism > phenotype (biochemical and external)
Genomics: Study of genomes of organisms; sequencing, mapping and gene interactions.
Proteomics: Study of complement of proteins in a system of an organism.
Transcriptomics: All expressed mRNA or transcripts (ncRNAs) made in one cell or a population of cells.
Metabolomics: Complete map of all small molecule metabolites in the human body or any other systems.
Structural genomics: Determination of 1D, 2D and 3D structure of all proteins in a given organisms.
Informatics: Application of information technology to the field of molecular biology; this information technology in specialized form is called ‘Computational Molecular Biology’, which deals with molecular aspects of biological systems..
Pharmacogenomics: Identification of genetic basis of heritable and inter-individual variation in response drugs.
Regulomics: Transcriptional factors and other molecules involved in regulation of Gene Expression
Gene expression:
Expression of housekeeping genes, as described in the earlier chapter is the most simplistic in terms of mechanism. Genes with specific promoter elements attract the enzyme, which in association with specific sigma factor identifies a specific promoter element and positions the enzyme in such a way, the enzyme tightly binds and initiates transcription exactly at start site. In these genes RNAP perse is incapable of initiating transcription, though it is properly positioned on the DNA; it requires correct sigma factor to activate initiation of specific gene. Many times, activation requires an activation factor; this kind of regulation is positive regulation, where the enzyme is activated by a factor.
Cells also respond, to changes at intracellular or extra cellular level (environment), by expressing required genes, which are hitherto remained silent and inactive. In the case of such genes, though the enzyme complex with its ubiquitous or specific sigma factor bound to the promoter, the enzyme is blocked of its progression by another protein called repressor. The repressor has to be made inactive or it has to be induced to dissociate, or the repressor should made into an activator to make the RNA-pol to initiate transcription, such expression is called negative regulation and positive regulation (latter). An interesting case is that a repressor becomes an activator. Some genes are regulated by both positive and negative regulatory elements. There are many regulatory mechanisms operated by a variety of factors and variety of modes. Some examples given below are only typical ones.
· A temporal change operates in the gene expression e.g. During endospores formation in B. subtilis.
· Regulation of lytic and lysogenic cycles of lambda phage.
· Expression of genes for the synthesis of certain metabolites to the needed for the cell.
· Induction or repression in response to.
· Coupled expression of topoisomerase to the needs of cell requirements.
· Expression of repair enzymes in response to DNA damage.
· Modification of nucleotides can change the expressional behavior of certain genes by methylation or demethylation of DNA at certain positions.
Some of the promoter sequences:
Whatever may be the intrinsic articulations, factors and mechanisms involved, they all have to take place at an identified locus where enzyme complex is positioned (a defined locus in the promoter). Then by various intricate modulations or mechanisms the enzyme complex is made to initiate transcription or an active enzyme is made inactive or not allowed to transcribe. In this regard, understanding of promoter elements, proteins and other factors, involved in the expression of typical genes is important.
Most of the genes in E. coli (more than ~4800), are organized into clusters and each of the clusters is controlled by the same promoter/operator elements. Such clusters are called Operons; there are 670-700 such operons in E. coli. Many operons are regulated by another group of factors called Regulons.
Promoter Elements of Some Genes:
|
Gene |
-35 |
N |
-10 |
N |
Start+1 |
|
Lac Z |
TTTACA |
N17 |
TATGTT |
N5/6 |
A/ G |
|
Lac I |
GGCGCA |
|
CATGAT |
N7 |
G |
|
Trp |
TTGACA |
N17 |
TTAACT |
N8 |
A |
|
Lambda |
TTGACA |
|
TATAAT |
N8 |
A/G |
|
Ara BAD |
TGACGC CTGACG |
N18 |
TACTGT |
N8 |
A |
|
Ara C |
TAGACA |
|
TGTCAT |
N7 |
G |
|
Rec A |
TTGATA (TAGACA) |
N18 |
TGTCAT |
N7 |
A |
|
Gal-P2 |
GTCACA |
|
TATGCT |
N7 |
A |
|
tRNA tyr |
TTTACA |
|
TATGAT |
N7 |
C G T |
|
Gro E |
TTTC5TTGAA |
N13 GC rich |
C4ATTTCTCTGGT |
|
C A C |
|
Dna K |
TCTC5TTGAT |
13 |
C4AT4AGTAGT |
|
C A A |
|
Sig 70 |
TGCCACCCTTGA |
15 |
GACGATATAGCAG |
|
T A A |
|
Rrn D1 |
GATC A7TAC |
12 |
TATAAT |
7 |
C G T |
|
Rrn X1 |
ATGCAT5CCGC |
13 |
TATAAT |
7 |
C A T |
|
Rrn A1 |
TTTAAATTTCCTC |
7 |
TATAAT |
7 |
C A C |
|
Rrn A2 |
GCA5TAATGC |
5 |
TACGAT |
7 |
A |
|
Lambda Pl (N) |
TTGACA |
|
GATACT |
5 |
C a t |
|
T7 |
|
|
TACG A CTCACTA |
|
A G G |
|
P 22ant |
TTGACA |
|
TATA |
|
|
|
UV 5 |
TTTACA |
|
TATAAT |
5 |
G A A |
A list of Proteins as Regulators:
|
Factor |
Mol.wt (KD) |
Effect |
Function |
|
Lac operon repressor |
37-38kd (tetramer) |
Blocks transcription |
Blocks the progression of RNAP |
|
Gal4 repressor/activator |
86-92 |
Blocks transcription |
Prevents the synthesis of Galactose utilizing enzymes when Galactose is absent |
|
Lex A repressor |
22.2 |
Blocks |
Prevents the expression DNA repair enzymes until repair
|
|
Trp repressor |
12.12.8kd(dimer) OR 20-22 |
Blocks |
Blocks transcription when trp present |
|
Lambda repressor cI |
27kDA |
Blocks Pl and PR |
Prevents the synthesis of protein required for lytic growth |
|
Lambda repressor |
|
Activates promoter PRM |
Increases the synthesis of CI by stimulating its own synthesis |
|
Lambda C II |
|
Activates transcription from PRE |
Stimulate the synthesis of proteins for lysogeny |
|
cAMP receptor protein
|
22.5kd (dimer) |
Activates prot III
Can block its own synthesis when glucose is absent |
Increase the synthesis of enzymes for utilizing other sugars in the absence of glucose |
|
Ara C |
42.6kd |
Activator form |
Increase the synthesis of enzymes for utilizing Arabinose when glucose is absent |
|
Ara C |
42.6 |
Repressor |
Blocks the synthesis of Arabinose utilizing enzymes when glucose is present |
|
Histidine |
54kda |
|
|
|
Hut repressor
|
27.6 |
Blocks |
Expression of two operons required for utilizing Histidine |
|
Gln. synthase A |
30 |
Activates |
Hut gene expression |
|
NTR-C (P) |
|
Activates |
Activates Gln.A synthase gene |
|
NTR-B(p) |
38.4 |
Activates |
Activates NTRP-C by protein kinase activity |
|
Spo-OA (B.subtilis)
|
|
Activates |
1.using sig 43 activates gene for sig F 2.using sig H activates gene for sig E |
|
GCN4 |
92 |
Activates GAL4 gene expression |
|
|
Lambda cIII |
17kd |
Provides stability to cIII |
|
|
|
|
|
|
Models of elementary gene circuits: Design of gene circuits: lessons from bacteria:
Michael E. Wall, William S. Hlavacek & Michael A. Sauvageau.
Inducible and repressible gene circuits have been modeled by considering the following processes: transcription and decay of regulator (r) and effector (e) mRNA; translation and dilution of regulator (R), which is a transcription factor (TF), and enzyme (E); and processes that influence the level of intracellular signal (S): reaction of substrates (X); transport of extracellular signal (S'); and degradation/dilution. Regulatory influences are indicated by arrows that terminate in the middle of another arrow. For inducible circuits, the effect of enzyme on degradation of signal is represented by a dashed arrow. For repressible circuits, transport of extracellular signal and feedback inhibition of catalysis are represented as dotted arrows.
System features can be defined with reference to the model:
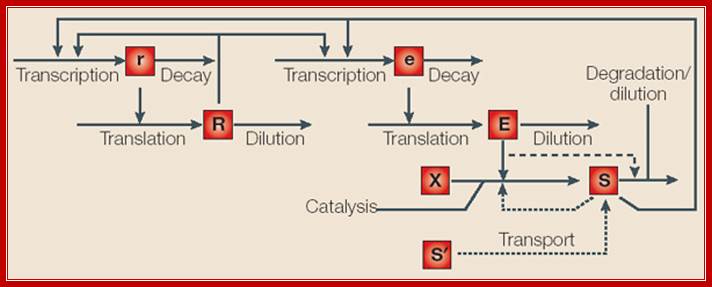
Design of gene circuits: lessons from bacteria; Inducible and repressible gene circuits have been modelled by considering the following processes: transcription and decay of regulator (r) and effector (e) mRNA; translation and dilution of regulator (R), which is a transcription factor (TF), and enzyme (E); and processes that influence the level of intracellular signal (S): reaction of substrates (X); transport of extracellular signal (S'); and degradation/dilution. Regulatory influences are indicated by arrows that terminate in the middle of another arrow. For inducible circuits, the effect of enzyme on degradation of signal is represented by a dashed arrow. For repressible circuits, transport of extracellular signal and feedback inhibition of catalysis are represented as dotted arrows. System features can be defined with reference to the model Michael E. Wall et al; Nature reviews
In systems with an activator (repressor) mode of control, the regulator exerts a positive (negative) influence on transcription of effector mRNA.
In systems with positive (negative) auto regulation, the regulator exerts a positive (negative) influence on transcription of regulator mRNA. The regulator might also have no influence on transcription of regulator mRNA.
In inducible (repressible) systems, an increase in the steady-state level of intracellular signal leads to an increase (decrease) in the steady-state level of enzyme.
In directly (inversely) coupled systems, an increase in the steady-state level of intracellular signal leads to an increase or decrease in the steady-state level of regulator. In uncoupled systems, changes in the steady-state level of intracellular signal have no effect on the steady-state level of regulator.
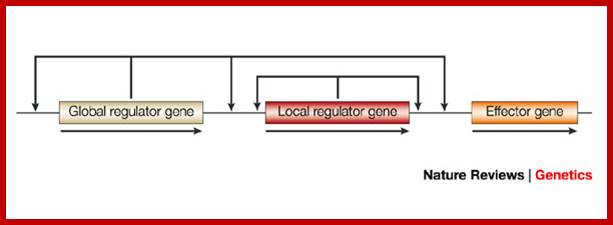
Transcriptional regulatory interactions in a gene circuit with a global and a local transcription factor. Design of gene circuits: lessons from bacteria; Each arrow begins at a transcription factor (TF) gene and terminates at the promoter of a gene, transcription of which might be regulated by the TF (only the TF interactions that regulate effector gene expression are necessarily present). The illustration of the gene circuit shown here is similar to that of the elementary gene circuit shown in Fig. 1. Analysis of the functional consequences of alternative designs requires a model at the level of detail shown in Box 3, including both the local and global signals. Michael E. Wall, William S. Hlavacek & Michael A. Savageau; http://www.nature.com
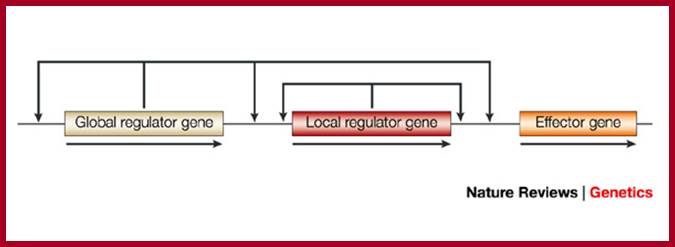
Design of gene circuits: lessons from bacteria; Each arrow begins at a transcription factor (TF) gene and terminates at the promoter of a gene, transcription of which might be regulated by the TF (only the TF interactions that regulate effector gene expression are necessarily present). The illustration of the gene circuit shown here is similar to that of the elementary gene circuit shown in Fig. 1. Analysis of the functional consequences of alternative designs requires a model at the level of detail shown in Box 3, including both the local and global signals. Michael E. Wall, William S. Hlavacek & Michael A. Savageau; http://www.nature.com
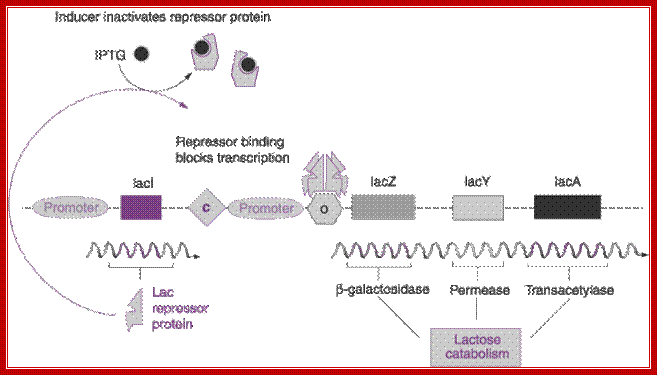
Regulation Lac operon: http://hi.baidu.com/
Science, Technology, and Mathematics;
E. coli genome codes for ~4488 proteins, 670-700 operons? ~27-337 pseudo genes (Yersinia pestis).
Operons:
Regulating a cluster of conjoint cistrons by common regulatory elements is called Operon. Estimates of E. coli data base indicate there are approximately 836-849- monocistronic genes, 1355-1455 polycistronic operons. There are 20 monocistronic and 57 polycistronic tRNA operons and six and seven polycistronic rRNA and ribosomal protein subunits operons.
Regulons:
- There are estimated ~173 regulons. In cell biology a regulon is a collection of operons under regulation by the same regulatory protein (s). This term is generally used for prokaryotic systems, for example quorum sensing in bacteria. It is a group of operons/genes spread around the chromosome but controlled by a common factor or stimulus; ; this can also be referred to Stimulons. http://en.wikipedia.org/wiki/Regulon
A bacterial regulon is a group of operons that are transcriptionally co-regulated by the same regulatory machinery, consisting of trans regulators (transcription factors or simply TFs) and cis regulatory binding elements in the promoters of the operons they regulate ( Han Zhang equal contributor, equal contributor
X
- Yanbin Yin equal contributor, equal contributor
X
- Victor Olman,
X
- Ying
Xu); some are local and some are global.
The genes of a regulon share a common regulatory element binding site or promoter. The genes comprising a regulon may be located non-contiguously in the genome. Ex. Crp regulon contains 230 operons, sigma S controls ~20 regulon, Copper ArcB/A regulon FNR regulon etc.
Very often different operons required for the same pathway or related pathways are controlled by the same protein or proteins. Such a system is also called Regulon. There are 173 well known Regulons in E.coli. en.wiktionary.org/wiki/regulon
- Modulons: Very often multiple Regulons are regulated by additional regulatory elements called Modulons. A modulon is a regulon concerned with multiple pathways or functions, in which operons may be under individual controls as well as common, pleiotropic regulatory protein. There are 100-150 modulons, ex. Red Ox moduon, Fur modulons; some can be nutrient and energy supply modulons, stress response modulons and cell-cell interacting modulons, polyamine modulon etc.
For example: the CAP modulon contains all regulons/operons, such as the lac operon and Ara regulon; that are regulated by CAP/cAMP, but each operon has other regulators as well.
Stimulons: Stimulation is a group of operons by a single stimulus is called Stimulon. Definitions of Stimulon on the Web:
- In
cell biology a stimulon is a collection of genes (which may be in operons
and regulons) regulated by a stimulus. This term is generally used for
prokaryotic systems, for example quorum sensing in bacteria, en.wikipedia.org/wiki/Stimulon. A system of genes that are
regulated by the same stimulus.
- All
the genes and gene clusters that are expressed in response to a distinct
physicochemical input, regardless of the specific regulators that mediate
such a response. Typically, one stimulon involves the action of more than
one transcription factor.
www.nature.com/nrmicro/journal/v3/n2/glossary/nrmicro1084_glossary.htm
Regulogs: Regulog is a set of co-regulated genes for which the regulatory sequence has been conserved across multiple organisms. Regulogger produces Regulogs.
Reg-Precise: A database of curated genomic inferences of transcriptional regulatory interactions in prokaryotes. The Reg-Precise is a database for capturing, visualization and analysis of transcription factor regulons that were reconstructed by the comparative genomic approach in a wide variety of prokaryotic genomes.
Yeast RRB Regulon- EBP2 is a member:
Christopher Wade,1 Kathleen A. Shea,1 Roderic k V. Jensen,2 and Michael A. McAlear1,*
Transcriptionally coregulated set of Genes are required for Ribosome and rRNA biosynthesis. In an effort to identify sets of yeast genes that are coregulated across various cellular transitions, gene expression data sets derived from yeast cells progressing through the cell cycle, sporulation, and diauxic shift were analyzed. A partitioning algorithm was used to divide each data set into 24 clusters of similar expression profiles, and the membership of the clusters was compared across the three experiments. A single cluster of 189 genes from the cell cycle experiment was found to share 65 genes with a cluster of 159 genes from the sporulation data set. Many of these genes were found to be clustered in the diauxic-shift experiment as well. The overlapping set was enriched for genes required for rRNA biosynthesis and included genes encoding RNA helicases, subunits of RNA polymerases I and III, and rRNA processing factors. A subset of the 65 genes was tested for expression by a quantitative-relative reverse transcriptase PCR technique, and they were found to be coregulated after release from alpha factor arrest, heat shock, and Tunicamycin treatment. Promoter scanning analysis revealed that the 65 genes within this ribosome and rRNA biosynthesis (RRB) regulon were enriched for two motifs: the 13-base GCGATGAGATGAG and the 11-base TGAAAAATTTT consensus sequences. Both motifs were found to be important for promoting gene expression after release from alpha factor arrest in a test rRNA processing gene (EBP2), which suggests that these consensus sequences may function broadly in the regulation of a set of genes required for ribosome and rRNA biosynthesis
eIF4E is a Central Node of an RNA Regulon that Governs Cellular Proliferation: Correspondence to Katherine L.B. Borden: katherine.borden@umontreal.ca
This study demonstrates that the eukaryotic translation initiation factor eIF4E is a critical node in an RNA regulon that impacts nearly every stage of cell cycle progression. Specifically, eIF4E coordinately promotes the messenger RNA (mRNA) export of several genes involved in the cell cycle. A common feature of these mRNAs is a structurally conserved, 50-nucleotide element in the 3′ untranslated region denoted as an eIF4E sensitivity element. This element is sufficient for localization of capped mRNAs to eIF4E nuclear bodies, formation of eIF4E-specific ribonucleoproteins in the nucleus, and eIF4E-dependent mRNA export. The roles of eIF4E in translation and mRNA export are distinct, as they rely on different mRNA elements. Furthermore, eIF4E-dependent mRNA export is independent of ongoing RNA or protein synthesis. Unlike the NXF1-mediated export of bulk mRNAs, eIF4E-dependent mRNA export is CRM1 dependent. Finally, the growth-suppressive promyelocytic leukemia protein (PML) inhibits this RNA regulon. These data provide novel perspectives into the proliferative and oncogenic properties of eIF4E.
Environmental Regulons:
Genes
can be regulated together even though they are located at different parts of
the bacterial chromosome. A group of genes regulated by the same
environmental signal is a regulon. An example is the rpoS
starvation regulon.
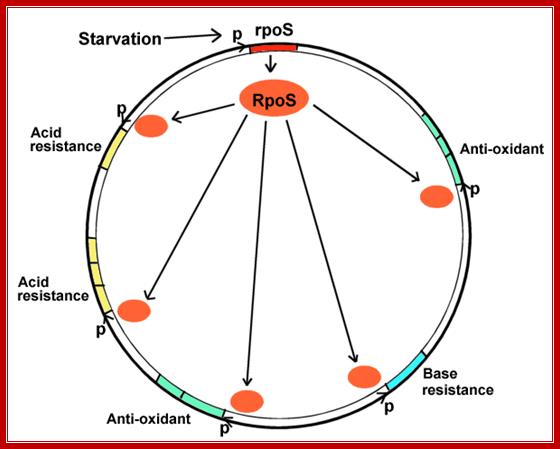
This diagram represents a Regulon where RpoS regulates very many genes. http://biology.kenyon.edu/
When bacteria enter digestive tract, how do they adjust to one’s body's defense mechanisms such as acid and antioxidant stress and others? When bacteria exit the intestine how do they cope with starvation?
- When bacteria use up their carbon sources, they express RpoS, the starvation sigma factor.
- RpoS joins RNA polymerase to initiate transcription of different environmental stress genes--genes protecting against all the different stresses that the bacteria might encounter before they enter a new human intestine. This phenomenon is known as cross-protection.
- The stress genes can be used for conditions as unrelated as acid or base resistance.
- The stress genes activated may or may not be part of multi-gene operons. They may face in opposite directions, from many different promoters, at all different loci around the genomic map.
‘Bacteria are the best genetic system to study genetic regulons. However, stress genes discovered in bacteria have been shown to have homologs in eukaryotic systems. For example, heat shock genes have been found in all organisms, including humans. Heat shock genes in humans also show cross-protection; for example, one heat shock protein interacts with the progesterone receptor and the contraceptive drug RU486.’
Control of RpoS in Global Gene Expression of Escherichia coli in Minimal Media:
RpoS, an alternative sigma factor, is critical for stress response in Escherichia coli. The RpoS regulon expression has been well characterized in rich media that support fast growth and high growth yields. In contrast, though RpoS levels are high in minimal media, how RpoS functions under such conditions has not been clearly resolved. In this study, we compared the global transcriptional profiles of wild type and an rpoS mutant of E. coli grown in glucose minimal media using microarray analyses. The expression of over 200 genes was altered by loss of RpoS in exponential and stationary phases, with only 48 genes common to both conditions. The nature of the RpoS-controlled regulon in minimal media was substantially different from that expressed in rich media. Specifically, the expression of many genes encoding regulatory factors (e.g., hfq, csrA, and rpoE) and genes in metabolic pathways (e.g., lysA, lysC, and hisD) were regulated by RpoS in minimal media. In early exponential phase, protein levels of RpoS in minimal media were much higher than that in Luria-Bertani media, which may at least partly account for the observed difference in the expression of RpoS-controlled genes. Expression of genes required for flagellar function and chemotaxis was elevated in the rpoS mutant. Western blot analyses show that the flagella sigma factor FliA was expressed much higher in rpoS mutants than in WT in all phase of growth. Consistent with this, the motility of rpoS mutants was enhanced relative to WT. In conclusion, RpoS and its controlled regulators form a complex regulatory network that mediates the expression of a large regulon in minimal media; (Annie Kolb akolb@pasteur.fr).
The σS factor of RNA polymerase:
RNA polymerase is responsible for the first step of gene expression and the target of numerous regulations. The bacterial holoenzyme consists of the core enzyme E and a sigma factor that enables the specific recognition of promoters and transcription initiation. The Enterobacteriaceae Escherichia coli and Salmonella enterica have seven sigma factors which compete for core binding. σ70, the "house-keeping" sigma factor, controls the transcription of all the essential functions of the bacteria. As cells reach the end of the exponential phase or are subjected to various environmental stresses, σS, also called σ38 comes into play. σS is responsible for the transcription of a few hundred genes, important for acquisition of the generalized stress resistance status and for survival in the stationary phase. σS is involved in resistance to osmotic, acid and oxidative stresses, biofilm formation and expression of virulence factors. Up to now, the exact function of nearly half of the genes regulated by σS is still unknown.
Functional Analysis of The Genes of The rpoS Regulon in Salmonella: (F. Norel, V. Robbe-Saule)
σS is essential for virulence and persistence of the facultative intracellular pathogen Salmonella enterica serotype Typhimurium which causes severe infections in man and animals. Our goal is to analyze the functions of the rpoS regulon in the physiology and pathogenesis of Salmonella and to identify the key functions of the regulon important for its survival in the environment, including its hosts. To perform a functional analysis of the rpoS regulon in Salmonella, we isolated σS-activated lacZ gene fusions from a bank of S. Typhimurium mutants. One-third of the fusions mapped to Salmonella DNA regions not present in E. coli K-12. This suggests that the composition of the rpoS regulon differs markedly in the two species. Most of the fusions mapped in genes of unidentified function. One of these, the katN gene, was shown to encode a non-haem catalase. katN is not present in E. coli K-12 but is conserved in entero-hemorrhagic E. coli O157, Klebsiella pneumoniae and Pseudomonas aeruginosa. Most of the remaining genes are likely to possess metabolic functions involved in adaptation of Enterobacteria under suboptimal growth conditions in natural environments.
LexA protein controls the expression of 43 genes [Courcelle et al.) (2001)
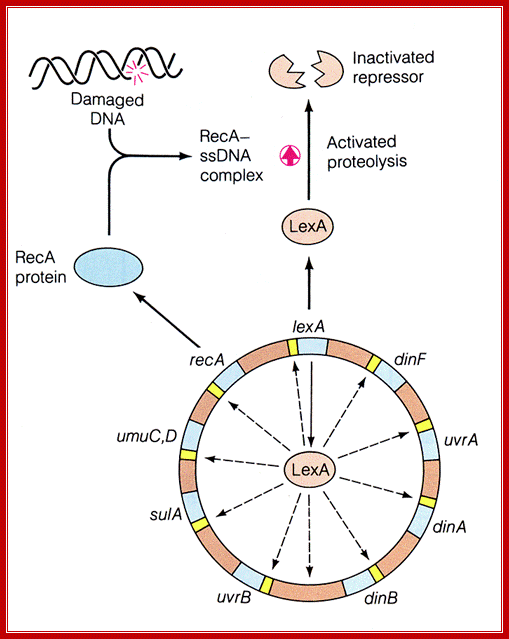
Lex A protein regulates ten genes; http://www.studyblue.com/
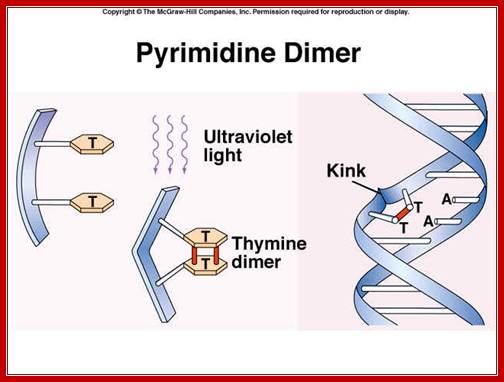
Pyrimidine dimers: http://faculty.southwest.tn.edu/
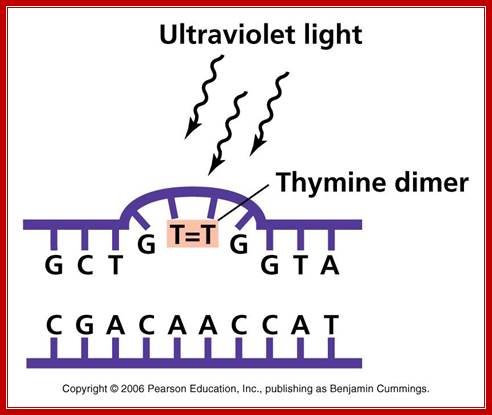
UV effect on nucleotides to generate Thymine dimers. http://academic.pgcc.edu
Role of LexA:
1) LexA protein binds to specific regions (SOS boxes) of DNA, represses expression of RecA gene and of the LexA gene too (need some RecA protein at all times) plus other genes (both those in the SOS system and others - about 20 genes in all, Chi (Χ) hotspot increases the derepression - by increasing RecA binding?). SOS promoters are destabilized by super helical stresses.

http://www.regensci.org/
2) With massive DNA injury, the products of that injury (ss DNA?) activate the RecA protein to phosphorylate itself to RecA~P, which polymerizes to form a fiber, RecA*.
3) RecA* + LexA ----> cleavage of LexA; can no longer bind to inhibit RecA synthesis ----> high level of RecA. RecA has two binding sites, one for ds and one for ss DNA.
4) RecA, LexA and other genes (including many DNA repair genes: RecB, RecC, UvrA etc.) turned on. LexA derepressed, but LexA protein inactivated by high level of RecA protein. UmuC, UmuD are required to replicate past unrepaired DNA lesions {pol II reinitiates DNA sysntesis at stalled forks after the lesion, leaving a gap which can be filled in properly by homologous recombination repair, 30-45 minutes after this - in slow growing cultures, the RecA system is begun}. The RecA, UmuC, UmuD system is activated by the cleavage of UmuD to UmuD', while it is still in contact with RecA*. A (UmuD')2UmuC complex is formed, which is a DNA polymerase (pol V) and can read through any unresolved template gaps, often replacing them with mismatched bases. (error prone repair); some of the enzymes in these systems are related. Further, DNA pol V is known to bind to DNA pol III subunits.
5) DNA repaired, RecA protein no longer phosphorylated (signal is gone), high level of derepressed LexA A gene makes much LexA protein; shuts off SOS system.
Recombination Systems:
(All require ATP hydrolysis)
|
RecBCD |
RecE |
Conjugational RecF |
++ |
|
- |
sbcA (Rac-20) mutation |
sbcB ( exo I), sbcC (nuclease with sbc D;recombination) mutations |
- |
|
RecA |
RecA |
RecA (binds well to poly (CA) (GT) as in Χ) |
RecA |
|
RecB |
- |
- |
RecB |
|
RecC |
- |
- |
RecC |
|
RecD |
- |
- |
- |
|
- |
RecE (Exo VIII) |
? |
- |
|
- |
RecF |
? |
|
|
- |
RecJ (exonuclease, ssDNA) |
RecJ |
? |
|
- |
- |
RecN (rad 8) |
? |
|
- |
RecO |
RecO |
? |
|
- |
RecQ |
RecQ (helicase?) |
? |
|
- |
- |
Ruv C (resolves Holiday junctions) |
? |
|
Gyrase |
? |
Uvr D (Mut U) (helicase?) |
? |
|
SSB |
- |
- |
? |
|
DNA PolI |
? |
Mut L (mismatch repair) |
? |
|
DNA Ligase |
? |
? |
? |
RecBCD System:
RecBCD system uses ds DNA end breaks, is like eukaryotic meiotic recombination. It acts to unwind DNA, to form large ss loops and uses ATP. When loop (either one) reaches Chi (C) recombinational hotspot ('5-GCTGGTGG-3'), the RecBCD enzyme cleaves it. This leaves a long ssDNA strand which the RecA protein and SSBs can act on to promote recombination. The RecBCD protein also has nuclease activity.
The maximum UV germicidal effect coincides with the peak absorbance of DNA (near 260nm) due to the dimerization of two adjacent thymines. E. coli cells have a system that recovers from DNA damage when it occurs. The best studied transcriptional response to DNA damage is the SOS response [Friedberg et al 1995, Walker et al. 1996].
This system can be divided into two classes: the SOS Photo reactivation repair and the SOS respond triggered by RecA protein. The first uses the photolyase, a poorly expressed enzyme (encoded by genes phrA and phrB) which binds the pyrimidine dimers and uses the blue light to split them apart as showed in Figure 13. Otherwise, single stranded DNA produced by several DNA-damaging agents can be bound by RecA protein, resulting in conversion of this protein to its activated form. The RecA repair system doesn’t need light and Lex A protein controls the expression of 43 genes [Courcelle et al] (2001) that cooperate together to repair the extensive genetic damage (Figure 14). RecA and LexA proteins play an important role for the regulatory of SOS Recombination System. A LexA binding site is present in all the SOS promoters genes' and it works as a repressor of SOS system. In presence of DNA damage (DNA Single Strains) RecA becomes active and interacts with LexA protein, the repressor of the SOS genes [Wagner et al., 1999]. This interaction triggers the autocatalytic cleavage of LexA and consequent destruction of its ability to function as a repressor, which results in the derepression of SOS genes [Mustard and Little 2000; Fernandez De Henestrosa et al. 2000]. When the damage is repaired, DNA single strands are not present in the cell and the RecA protein no longer promotes the auto-cleavage of the LexA which is restored to its initial repression level.
|
|
|
Figure: Autogenous control of LexA transcription determines many SOS response properties. To study these properties we have constructed a non-autogenous circuit (a). Camas et al. 2006. PNAS USA. 103: 12718–12723; Biehl A, et al; Abteilung für Pflanzenzüchtung und Ertragsphysiologie, Germany
|

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Analysis of 101 nuclear transcriptomes reveals 23 distinct regulons and their relationship to metabolism, chromosomal gene distribution and co-ordination of nuclear and plastid gene expression.
Abstract
Post-endosymbiotic evolution of the proto-chloroplast was characterized by gene transfer to the nucleus. Hence, most chloroplast proteins are nuclear-encoded and the regulation of chloroplast functions includes nuclear transcriptional control. The expression profiles of 3292 nuclear Arabidopsis genes, most of them encoding chloroplast proteins, were determined from 101 different conditions and have been deposited at the GEO database (http://www.ncbi.nih.gov/geo/). The 1590 most-regulated genes fell into 23 distinct groups of co-regulated genes (regulons). Genes of some regulons are not evenly distributed among the five Arabidopsis chromosomes and pairs of adjacent, co-expressed genes exist. Except regulons 1 and 2, regulons are heterogeneous and consist of genes coding for proteins with different subcellular locations or contributing to several biochemical functions. This implies that different organelles and/or metabolic pathways are co-ordinated at the nuclear transcriptional level, and a prototype for this is regulon 12 which contains genes with functions in amino acid and carbohydrate metabolism, as well as genes associated with transport or transcription. The co-expression of nuclear genes coding for subunits of the photosystems or encoding proteins involved in the transcription/translation of plastome genes (particularly ribosome polypeptides) (regulons 1 and 2, respectively) implies the existence of a novel mechanism that co-ordinates plastid and nuclear gene expression and involves nuclear control of plastid ribosome abundance. The co-regulation of genes for photosystem and plastid ribosome proteins escapes a previously described general control of nuclear chloroplast proteins imposed by a transcriptional master switch, highlighting a mode of transcriptional regulation of photosynthesis which is different compared to other chloroplast functions. From the evolutionary standpoint, the results provided indicate that functional integration of the proto-chloroplast into the eukaryotic cell was associated with the establishment of different layers of nuclear transcriptional control.
Modulons: Very often multiple Regulons are regulated by additional regulatory elements called Modulons. A modulon is a regulon concerned with multiple pathways or functions, in which operons may be under individual controls as well as common, pleiotropic regulatory protein.
For example: the CAP modulon contains all regulons/operons, such as the lac operon and ara regulon, that are regulated by CAP/cAMP, but each operon has other regulators as well. This protein is annotated as belonging to Pfams PF00027 (cAMP/cGMP binding domain protein) and PF00325 (Crp family of bacterial transcriptional regulators). Crp family proteins are very diverse but may be allocated into subfamilies based on sequence and it includes Crp, Clp, CysR, FixK, Flp, Fnr, FnrN, HlyX and NtcA. They are supposed to bind DNA via their C-terminus HTH domain. Can we group this category of protein I Regulon or modulon; however it is global regulator of many genes and operons
Polyamine Modulon in Escherichia coli: Genes Involved in the Stimulation of Cell Growth by Polyamines .
Kazuei Igarashi* and Keiko Kashiwagi
Sciences, Chiba Institute of Science, 15-8 Shiomi-cho, Choshi, Chiba 288-0025 * To whom correspondence should be addressed. Phone: +81-43-226-2871, Fax: +81-43-226-2873, E-mail: iga16077@p.chiba-u.ac.jp
We
have
recently proposed an idea to explain how polyamines
enhance cell growth in Escherichia
coli. Since most polyamines exist
as
polyamine-RNA
complexes, our idea is that polyamines stimulate
several kinds of protein synthesis
which are important for cell growth at
the level of translation. We found that
synthesis of oligopeptide binding protein (OppA),
which is important for nutrient supply, adenylate
cyclase
(Cya),
RNA
polymerase ![]() 38 subunit (RpoS), transcription factor
of iron transport operon (FecI), and
transcription
factor
of growth-related genes including rRNA
and
some kinds of tRNA synthesis (Fis)
was
enhanced
by polyamines at the level of translation.
We proposed that a group of genes whose expression is
enhanced
by polyamines at the level of translation
be referred to as a "polyamine modulon."
By DNA microarray, we found that
309 of 2,742 mRNA species were up-regulated
by polyamines. Among the 309
up-regulated genes, transcriptional enhancement
of at
least
58 genes might be attributable to increased
levels of the transcription factors Cya,
RpoS, FecI, and Fis. This
unifying molecular mechanism is proposed to underlie the physiological
role of polyamines in controlling the growth of Escherichia
coli
38 subunit (RpoS), transcription factor
of iron transport operon (FecI), and
transcription
factor
of growth-related genes including rRNA
and
some kinds of tRNA synthesis (Fis)
was
enhanced
by polyamines at the level of translation.
We proposed that a group of genes whose expression is
enhanced
by polyamines at the level of translation
be referred to as a "polyamine modulon."
By DNA microarray, we found that
309 of 2,742 mRNA species were up-regulated
by polyamines. Among the 309
up-regulated genes, transcriptional enhancement
of at
least
58 genes might be attributable to increased
levels of the transcription factors Cya,
RpoS, FecI, and Fis. This
unifying molecular mechanism is proposed to underlie the physiological
role of polyamines in controlling the growth of Escherichia
coli
Stimulon: Stimulation of a group of operons by a single stimulus is called Stimulon. Definitions of Stimulon on the Web:
- In cell biology a stimulon is a collection of genes (which may be in operons and regulons) under regulation by the same stimulus. This term is generally used for prokaryotic systems, for example quorum sensing in bacteria.
- A
system of genes that are regulated by the same stimulus
en.wiktionary.org/wiki/stimulon - All
the genes and gene clusters that are expressed in response to a distinct
physicochemical input, regardless of the specific regulators that mediate
such a response. Typically, one stimulon involves the action of more than
one transcription factor.
www.nature.com/nrmicro/journal/v3/n2/glossary/nrmicro1084_glossary.html
The H2O2 Stimulon in Saccharomyces cerevisiae*
Christian Godon‡, Gilles Lagniel‡, Jaekwon Lee§, Jean-Marie Buhler‡, Sylvie Kieffer¶, Michel Perrot‖, Hélian Boucherie‖, Michel B. Toledano‡§ and Jean Labarre‡
The changes in gene expression underlying the yeast adaptive stress response to H2O2were analyzed by comparative two-dimensional gel electrophoresis of total cell proteins. The synthesis of at least 115 proteins is stimulated by H2O2, whereas 52 other proteins are repressed by this treatment. We have identified 71 of the stimulated and 44 of the repressed targets. The kinetics and dose-response parameters of the H2O2 genomic response were also analyzed. Identification of these proteins and their mapping into specific cellular processes give a distinct picture of the way in which yeast cells adapt to oxidative stress. As expected, H2O2-responsive targets include an important number of heat shock proteins and proteins with reactive oxygen intermediate scavenging activities. Exposure to H2O2 also results in a slowdown of protein biosynthetic processes and a stimulation of protein degradation pathways. Finally, the most remarkable result inferred from this study is the resetting of carbohydrate metabolism minutes after the exposure to H2O2. Carbohydrate fluxes are redirected to the regeneration of NADPH at the expense of glycolysis. This study represents the first genome-wide characterization of a H2O2-inducible stimulon in a eukaryote.
Aerobic organisms have to maintain a reduced cellular redox environment in the face of the pro-oxidative conditions’ characteristic of aerobic life. The incomplete reduction of oxygen to water during respiration leads to the formation of redox-active oxygen intermediates (ROI)1 such as the superoxide anion radical (O2 −), hydrogen peroxide (H2O2), and the hydroxyl radical (OH⋅) (for review. ROI are also produced during the β-oxidation of fatty acids, and upon exposure to radiation, light, metals, and redox active drugs. Oxidative stress results from abnormally high levels of ROI which perturb the cell redox status and leads to damage to lipids, proteins, DNA, and eventually cell death. Living organisms constantly sense and adapt to such redox perturbations by the induction of batteries of genes or stimulons whose products act to maintain the cellular redox environment. In Escherichia coli, two distinct stimulons exist, one for H2O2 and the other for O⨪2, each consisting of a set of 30–40 proteins (for review). The genes encoding nine of the H2O2-inducible proteins are controlled by the transcriptional regulator OxyR and include katG (catalase), ahpCF (an alkyl hydro peroxide reductase), gorA (glutathione reductase), and dps (a nonspecific DNA binding protein). The genes encoding nine of the O⨪2-inducible proteins are controlled by the SoxR/S transcriptional regulators and include soda (manganese superoxide dismutase), zwf1(glucose-6-phosphate dehydrogenase), nfo (DNA repair exonuclease IV), fumC (fumarase C), and micF (an antisense RNA regulator). Most of the remaining proteins of these stimulons are unknown, but their identification would increase our understanding of the mechanisms of cellular redox control and ROI metabolism. The yeast Saccharomyces cerevisiae can also adapt to both H2O2- and O⨪2-generating drugs by the induction of two distinct but overlapping stimulons for H2O2 and O. Yeast has the same defense mechanisms as higher eukaryotes and offers the power of genome-wide experimental approaches owing to the availability of the complete sequence of its genome. It therefore represents an ideal eukaryotic model in which to study the cellular redox control and ROI metabolism.
We recently established a general method to identify yeast proteins based on two-dimensional gel electrophoresis. We used this genome-wide experimental approach to characterize proteins whose expression is altered upon exposure to low doses of H2O2. Such an oxidative stress challenge results in a dramatic genomic response involving at least 167 proteins. Identification of these proteins and their mapping into cellular processes give a global view of the ubiquitous cellular changes elicited by H2O2 and provides the framework for understanding the mechanisms of cellular redox homeostasis and H2O2 metabolism.
A novel potassium Deficiency-Induced Stimulon in Anabaena torulosa. Alahari A, Apte SK. Bhabha Atomic Research Centre, Mumbai, India.
Potassium deficiency enhanced the synthesis of fifteen proteins in the nitrogen-fixing cyanobacterium Anabaena torulosa and of nine proteins in Escherichia coli. These were termed potassium deficiency-induced proteins or PDPs and constitute hitherto unknown potassium deficiency-induced stimulons.
Potassium deficiency also enhanced the synthesis of certain osmotic stress-induced proteins. Addition of K+ repressed the synthesis of a majority of the osmotic stress-induced proteins and of PDPs in these bacteria. These proteins contrast with the dinitrogenase reductase of A. torulosa and the glycine betaine-binding protein of E. coli, both of which were osmo-induced to a higher level in potassium-supplemented conditions. The data demonstrate the occurrence of novel potassium deficiency-induced stimulons and a wider role of K+ in regulation of gene expression and stress responses in bacteria
The glucose-starvation a Stimulon of Escherichia coli: Nyström T.; Department of General and Marine Microbiology, University of Göteborg, Sweden.
It is induced and repressed synthesis of enzymes of central metabolic pathways and role of acetyl phosphate in gene expression and starvation survival.
Proteins of the glucose-starvation stimulon were identified by using two-dimensional gel electrophoresis and the gene-protein database of Escherichia coli. Members of this stimulon included enzymes of the Emden-Meyerhof-Parnas (EMP) pathway, phosphotransacetylase (Pta) and acetate kinase (AckA) of the acetyl phosphate/acetate production pathway, and formate transacetylase. The synthesis of these enzymes was found to be induced concomitantly with the decreased synthesis of enzymes of the Krebs cycle. Thus, the modulation in the synthesis of specific proteins during aerobic glucose starvation is, in part, similar to the response of cells shifted to anaerobiosis.
These modulations suggest that the glucose-starved cell increases the relative flow of carbon through the Pta-AckA pathway. Indeed, the ability to synthesize acetyl phosphate, an intermediate of the pathway, appears to be indispensable for glucose-starved cells as pta and pta-AckA double mutants were found to be impaired in their ability to survive glucose starvation. The survival characteristics of AckA mutants and the wild-type parent were indistinguishable. Moreover, the Pta mutant failed to induce several proteins of the glucose-starvation stimulon.
Regulation of gene expression by small non-coding RNAs: a quantitative view:
Yishai Shimoni1, Gilgi Friedlander2, Guy Hetzroni1, Gali Niv2, Shoshy Altuvia2, Ofer Biham1 & Hanah Margalit:
The importance of post-transcriptional regulation by small non-coding RNAs has recently been recognized in both pro- and eukaryotes. Small RNAs (sRNAs) regulate gene expression post-transcriptionally by base pairing with the mRNA. Here we use dynamical simulations to characterize this regulation mode in comparison to transcriptional regulation mediated by protein–DNA interaction and to post-translational regulation achieved by protein–protein interaction. We show quantitatively that regulation by sRNA is advantageous when fast responses to external signals are needed, consistent with experimental data about its involvement in stress responses. Our analysis indicates that the half-life of the sRNA–mRNA complex and the ratio of their production rates determine the steady-state level of the target protein, suggesting that regulation by sRNA may provide fine-tuning of gene expression. We also describe the network of regulation by sRNA in Escherichia coli, and integrate it with the transcription regulation network, uncovering mixed regulatory circuits, such as mixed feed-forward loops. The integration of sRNAs in feed-forward loops provides tight repression, guaranteed by the combination of transcriptional and post-transcriptional regulations.
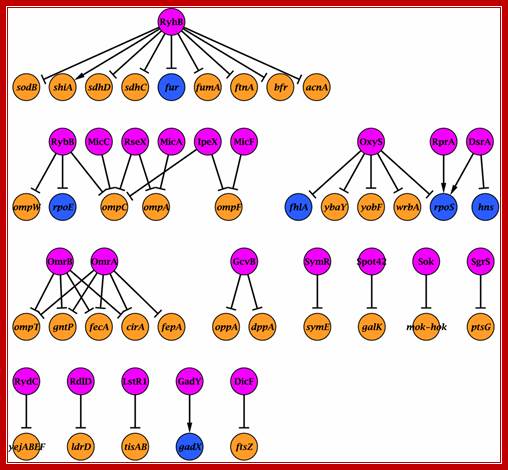
The sRNA–target network. Nodes represent sRNAs and their targets (see Supplementary information for references). sRNAs are in pink, protein-coding genes in orange and genes encoding transcriptional regulators in prokaryotes. http://msb.embopress.org/
A second paradigm for gene activation in bacteria: Buck M, Bose D, Burrows P, Cannon W, Joly N, Pape T, Rappas M, Schumacher J, Wigneshweraraj S, Zhang X. Faculty of Natural Sciences: Imperial College London, London SW7 2AZ, UK. m.buck@imperial.ac.uk.
Control of gene expression is key to development and adaptation. Using purified transcription components from bacteria, we employ structural and functional studies in an integrative manner to elaborate a detailed description of an obligatory step, the accessing of the DNA template, in gene expression. Our work focuses on a specialized molecular machinery that utilizes ATP hydrolysis to initiate DNA opening and permits a description of how the events triggered by ATP hydrolysis within a transcriptional activator can lead to DNA opening and transcription. The bacterial EBPs (enhancer binding proteins) that belong to the AAA (+) (ATPases associated with various cellular activities) protein family remodel the RNAP (RNA polymerase) holoenzyme containing the sigma (54) factor and convert the initial, transcriptionally silent promoter complex into a transcriptionally proficient open complex using transactions that reflect the use of ATP hydrolysis to establish different functional states of the EBP. A molecular switch within the model EBP we study [called PspF (phage shock protein F)] is evident, and functions to control the exposure of a solvent-accessible flexible loop that engages directly with the initial RNAP promoter complex. The sigma (54) factor then controls the conformational changes in the RNAP required to form the open promoter complex; m.buck@imperial.ac.uk.