Genetics of Apoptosis:
Genes and Mechanism:
Death Transducing Receptors:
Any factor, in any form that impinges on cells has to be first recognized by specific receptors and then the message has to be transduced to internal milieu to asses the signal and if needed execute molecular actions.
Receptors are cellular sentinels, located on the surface of the cell membranes and scan all the external signals that are impinged upon all the time. Thousand of such specific receptors to specific cell type are found located on their cell surface. They span the whole cell surface and they are found in different shapes with different structure and different functions.
Apoptotic receptors are varied and many, such as Fas-R (CD95), TNF-R1 and 2, Apo-1, rpr gene (Reaper in Drosophila), TRAIL1, TRAIL 2 (TNF related apoptosis inducing ligand receptors DR4 and DR5).
The receptors are located in plasma membrane with an external domain and a cytosolic domain and a transmembrane domain. For example when Fas-L ligand binds to Fas-R forms; it aggregates into trimers’ (actually dimer). When a specific signal, binds to a specific receptor on to external domain, it leads to certain conformation changes in the receptor proteins. This change is critical and the same is manifested at cytosolic side of the membrane. In the case of Fas-R binding leads to trimerization, it further induces more clustering at cytosolic surface.
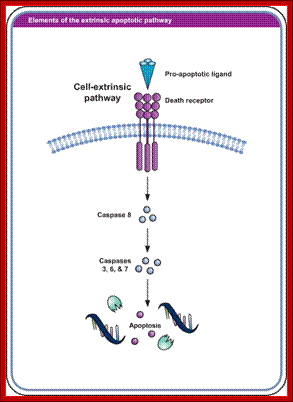
Extrinsic pathway for programmed cell death; http://www.biooncology.com/
The cytosolic domain interacts with respective cytoplasmic components and executes its actions there off. The cytosolic domain found at carboxy terminal of Fas-R and other related receptor proteins contain a 80 amino acid sequence, more or less same (28% conserved) among their related members. This region is called death domain or DD. Interaction between the death domains and cellular components results in activating a set of proteases.
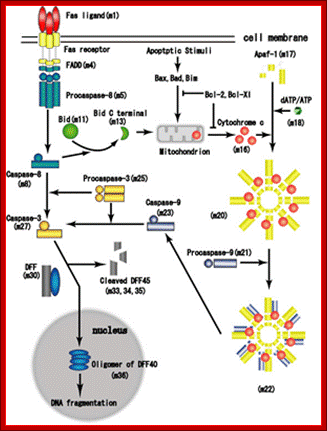
Proposed steps of apoptosis induced by Fas ligand: Fas ligands, which usually exist as trimmers, bind and activate their receptors by inducing receptor trimerization. Activated receptors recruit adaptor molecules such as Fas-associating protein with death domain (FADD), which recruit procaspase 8 to the receptor complex, where it undergoes autocatalytic activation. Activated caspase 8 activates caspase 3 through two pathways; The complex one is that caspase 8 cleaves Bcl-2 interacting protein (Bid) and its COOH-terminal part translocates to mitochondria where it triggers cytochrome c release. The released cytochrome C bind to apoplectic protease activating factor-1 (Apaf-1) together with dATP and procaspase 9 and activates caspase 9. The caspase 9 cleaves procaspase 3 and activates caspase 3. The other pathway is that caspase 8 cleaves procaspase3 directly and activates it. The caspase 3 cleaves DNA fragmentation factor (DFF) 45 in a heterodimeric factor of DFF40 and DFF45. Cleaved DFF45 dissociates from DFF40, inducing oligomerization of DFF40 that has DNase activity. The active DFF40 oligomer causes the internucleosomal DNA fragmentation, which is an apoptotic hallmark indicative of chromatin condensation. http://genomicobject.net/

Comparison between active and inactive forms of caspases. Newly produced caspases are inactive. Specifically cleaved caspases will dimerize and become active. https://sites.google.com
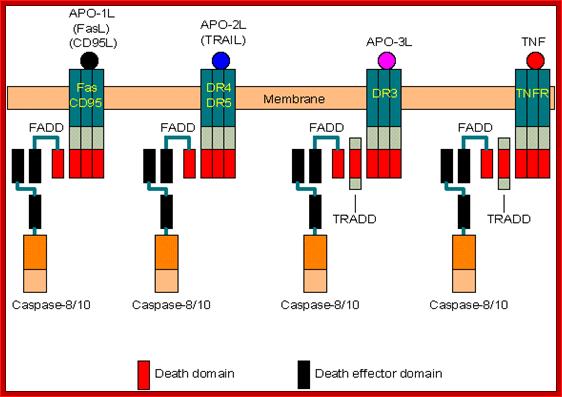
Transmembrane Signal receptors bound to specific Caspases
Coupling of caspase 8 or 10 to death receptors:
- Death receptors: Fas/CD95, DR4/DR5, DR3, and TNFR (Tumor Necrosis Factor Receptor).
- Adaptors: FADD (Fas-associated death domain protein) and TRADD (TNFR-associated death domain protein).
- Activation: Binding of death ligands (FasL/CD95L, TRAIL/APO-2L, APO-3L and TNF) induces trimerization of their receptors, which then recruit adaptors and activate caspases.
- Note: TRADD is involved only in the coupling between caspases and DR3 or TNFR. This adaptor can also recruit other proteins (web link) to inhibit apoptosis through the NF-kB pathway.
- As shown in the above figure, a variety of death ligands (FasL/CD95L, TRAIL, APO-3L and TNF) can induce apoptosis. It is natural to see if they can kill cancer cells without affecting normal cells. TNF was first investigated in the 1980s for cancer therapy, but with disappointing results. Then CD95L (FasL) was tested in the 1990s. The results were still not satisfactory. Recently, TRAIL has been demonstrated to be highly selective for transformed cells, with minimal effects on normal cells. It could be an effective drug for both cancer and AIDS. https://sites.google.com/
Fas ligand binding domain, after binding activates FasL Associated Death Domain (FADD) and activates Caspase enzymes:
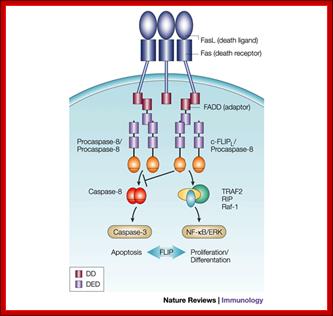
Regulation of death-receptor-induced gene
expression by FLIP: The long form of cellular FLIP (FLIPL) inhibits
complete processing and activation of caspase-8 at the receptor level and
thereby exerts an anti-apoptotic function. Moreover, FLIPL mediates
the recruitment/stabilization of additional signalling molecules, such as
tumour-necrosis factor receptor-associated factor 1 (TRAF1), TRAF2, receptor-interacting protein (RIP)
and Raf-1 at the death-inducing signalling complex, and might thereby have an
active role in the control of death receptor-mediated gene expression by means of the nuclear factor  B
(NF-B)
and extracellular signal-regulated kinase (ERK) pathways. (FLIP,
FLICE/caspase-8 inhibitory protein.); Margot
Thome & Jürg Tschopphttp://www.nature.com/; http://www.web-books.com/
B
(NF-B)
and extracellular signal-regulated kinase (ERK) pathways. (FLIP,
FLICE/caspase-8 inhibitory protein.); Margot
Thome & Jürg Tschopphttp://www.nature.com/; http://www.web-books.com/
![]()
The long form of cellular FLIP (FLIPL) inhibits
complete processing and activation of caspase-8 at the receptor level and
thereby exerts an anti-apoptotic function. Moreover, FLIPL mediates the recruitment/stabilization
of additional signaling molecules, such as tumour-necrosis factor
receptor-associated factor 1 (TRAF1), TRAF2,
receptor-interacting protein (RIP) and Raf-1 at the death-inducing signaling
complex, and might thereby have an active role in the control of death
receptor-mediated gene expression by means of the nuclear factor ![]() B
(NF-
B
(NF-![]() B)
and extracellular signal-regulated kinase (ERK)
pathways. (FLIP, FLICE/caspase-8 inhibitory protein.)
B)
and extracellular signal-regulated kinase (ERK)
pathways. (FLIP, FLICE/caspase-8 inhibitory protein.)
Regulation of Death –receptor-induce gene expression by FIP; Margot Thome & Jürg Tschopp; http://www.nature.com/
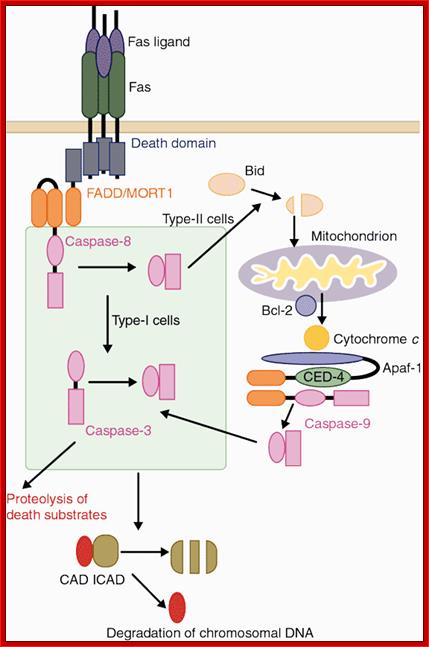
Two signaling pathways for Fas ligand (FasL)-induced apoptosis. Binding of FasL to Fas recruits procaspase-8 through the FADD adaptor, which results in processing of procaspase-8 into the active enzyme. In type-I cells such as thymocytes, caspase-8 directly cleaves caspase-3, which is an effector caspase. In type-II cells such as hepatocytes, caspase-8 cleaves Bid and the truncated Bid stimulates the release of cytochrome c from mitochondria. Cytochrome c, together with Apaf-1 and ATP, then activates caspase-9, which in turn then activates caspase-3. One of the substrates of caspase-3 is ICAD (inhibitor of caspase-activated DNase (CAD)). Cleavage of ICAD by caspase-3 activates CAD, which causes DNA degradation in nuclei. Shigekazu Nagata; http://www.nature.com/
Cell death related genes:
These are a number of genes involved in causing death. Among them proteases play an important role. Genetic studies in C.elegans (Ces), Drosophila and human beings resulted in identifying 14 cell death genes called Ced genes.
· Ces1, Ces2, Ces3-5 genes determine cell death specificity. Ces-1 causes death of 4 cells in pharynx. Ces 2 causes death of neuronal cells, but mutation in the gene prevents the death of few neurons. Each have their own specificity.
· Egl –egg laying, these cells die in males.
· Proteases, with Cysteine as an active site, that cleaves proteins at carboxyl side of aspartate residue, are called Caspases (Ced). Caspases are a family of 14 or more proteins; Ced1 to Ced 14. All of them are synthesized as pro-Caspases. Caspases are grouped into two, the first group consists of Caspases related to inflammation; such as casp-8, 9 & 10, they have long pro-domains and initiate cell death and the second group consists of casp-3, 6 &7 few more, they have short prodomains and cleave various structures. They are involved in responding to cell death signal, they are Ced 3 and Ced 6-10.
· Ced 3, Ced4, Ced8 and Ced9 control and execute death. Ced4 codes for a calcium binding protein.
· Ced3 is Cysteine protease; it is the most potent protease of all other Ced proteins. This has a homology with that of interleukin 1 b converting enzyme called ICE, which is also a Cysteine protease but required for the death of lymphocytes. Absence of this does not prevent apoptosis. Ced3 recognizes a sequence –YVAD (Tyrosine, Valine, Alanine, Aspartate) and ICE recognize DEVD sequence (Aspartate, Glutamate, Valine, Aspartate). Ced9 can protect the cell from death or it can be deadly. This has a homology with Bcl2. Ced 9 is also associated with membranes such as mitochondria, nucleus and ER.
· Ced1, Ced2, ced5, ced6, ced7 and Ced10 control the engulfment of the degraded cellular components by macrophages.
· Nuc1 is an endonuclease, controls degradation of engulfed matter by activated macrophages.
· Transcriptional activators- HLF, DBP and TEF/VBP. Transcriptional repressors-E4BP4, Giant, and Ces2.
· Bcl2 an oncogene, it is a member of multi protein complex; it has many related gene members. Bcl2 is a transmembrane protein localized in the outer membrane of mitochondria, nuclear membranes and endoplasmic membranes. Bax: it is associated with Bcl2 in membranes and causes the release of Cyt.C.
· Myc and p53, depending upon circumstances they cause survival of cells or induce the cells to death.
· BID, it is an inhibitor protein.
· APAF1, it is an apoptosis protease-activating factor.
· Cytochrome–C, it is mitochondrial protein; with the release of this protein from peri-mitochondrial space, it activates the whole process of cell death
· FADD: FAS-R associated death domain factor, associated with FAS-R. at cytosolic side.
· TRADD: TNFR α1 associated death domain factor, it is associated with TNF a1 receptor at cytosolic side.
|
Name |
KDs |
Target |
Activation |
Domain |
Substrates |
Function/s |
|
Caspase1, ICE |
45920+10 |
WHED |
TPLD, FED |
CARD |
Pro-IL, self |
Against inflammation |
|
Caspase2 |
48(18+12 |
DVAD |
DNKD |
CARD |
Self |
Initiates |
|
Caspase3 |
32(17+12) |
DMQD |
ESMD |
|
PARP, PK, self |
Initiator |
|
Caspase4 |
|
EHD, LEVD |
LEED |
CARD |
Pro-IL, self |
inflamation |
|
Caspase5 |
|
EHD |
WVRD, LEAD |
Self |
|
inflamation |
|
Caspase6 |
34 |
VEHD |
DVVD |
|
|
|
|
Caspase7 |
34(20+12) |
DEVD |
IQAD |
DED |
PARP, ICAD |
Effector |
|
Caspase8 |
53(18+11) |
ETD+ |
VET+ |
DED |
PARP, self |
Initiator |
|
Caspase9 |
50 |
LEHD |
PEPD |
CARD |
PARP, self |
Initiator |
|
Caspase10 |
55(17+12) |
IEHD |
IEHD |
DED |
Pocaspase3, 7 |
Initiates |
|
Caspase11-14 |
|
|
|
|
|
Swelling |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
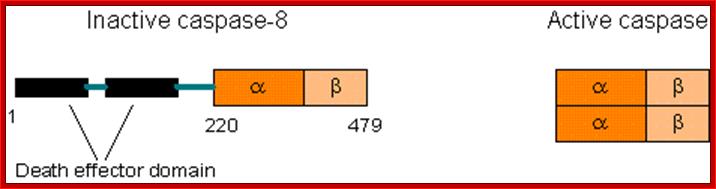
Comparison between active and inactive forms of caspases. Newly produced caspases are inactive. Specifically cleaved caspases will dimerize and become active; http://www.web-books.com/
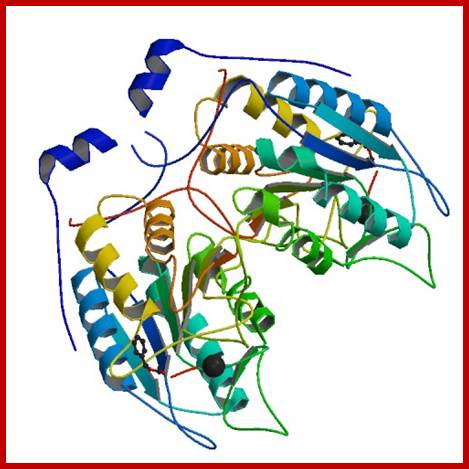
Human CASPASE 1 protein; http://www.rcsb.org;
Components involved in apoptosis:
Nucleus: Nuclear lamins, nucleoplasmins, SR proteins, hnRNPs, some Transcription factors such as RNA Pl (pstram) factors, MDM2, RB and inhibitors such as p27 and p21.
DNA: Repair enzymes, including RAD and others, PARP, Topoisomerases, and inhibitors of Caspases (iCAD/DFF45-Caspase activated DNase or DNA fragment factor).
Cytoskeleton structures: Actins, Gelsolins, and pectrins, Keratins.
Cytoplasmic components: beta catenins, Bcl2.
Protein kinases: DNA dependent protein kinase like ATM/ATR, Protein kinase C, CAM kinase, MAPs and ERK kinases, protein kinase-B, Raf1

The diagram shows what happens when cells are signaled by cell surviving signals; C Myc apoptotic pathway; www.learningame.org
Plant Cell Apoptosis II- This diagram is another showing the cellular changes when cell is prompted by cell survival signals, where Caspases are inactivated http://www.mdpi.com/;Daniela Cosentino-Gomes et al; http://www.mdpi.com/
Operation of Death program:
Signal from outside:
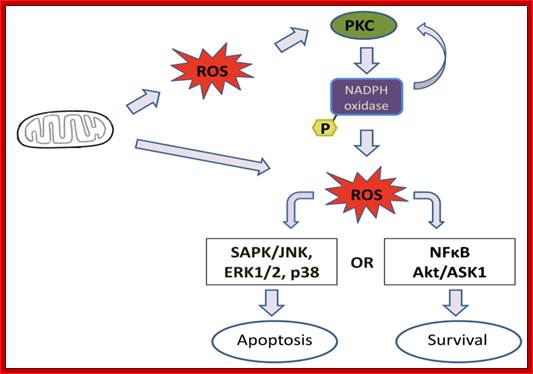
On binding of signal molecules to their respective receptors (signal transducers) such as Fas-R and TNFa1-R, receptor undergoes conformational changes and gets activated. This leads to activation of mitochondria which leads to release of Ca2+.
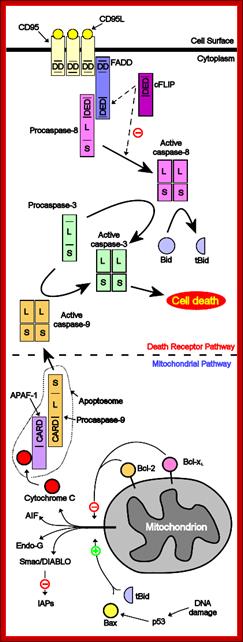
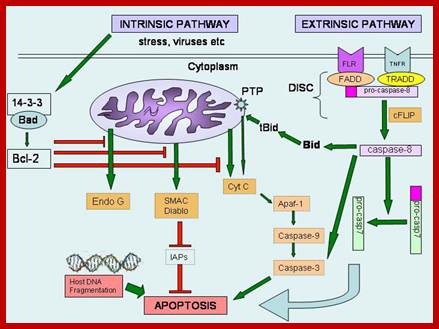
Sequential events from external signaling to mitochondrial release of Ca2+ ions, that leads to death process. Cell biology of diseases and exercise; http://pt851.wikidot.com
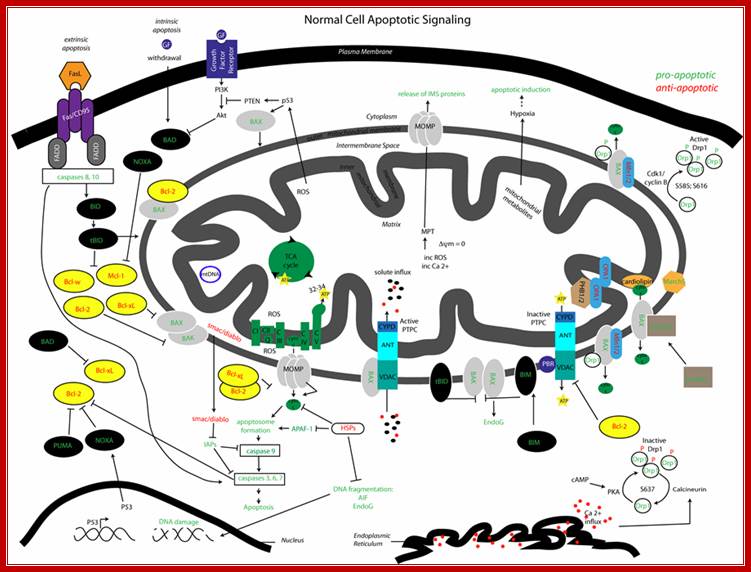
Mitochondrial pathways: interactions between apoptotic signaling and mitochondrial dynamics. Visualized is the complex array of proteins and signaling molecules that are involved in normal apoptotic signaling, both extrinsic and intrinsic, and the interaction of mitochondrial dynamics players that modify mitochondrial morphology and consequentially impact apoptotic regulation. Green colored text indicates pro-apoptotic signaling and red colored text indicates anti-apoptotic signaling. Emily R Roberts a, Kelly Jean Thomashttp://journals.sfu.ca/
A sensor of Calcium Uptake:
In cell's power plants mitochondria increase their energy production in response to calcium signals in the cytoplasm. A regulator of the elusive mitochondrial calcium channel has now been identified.
Calcium ions are transferred directly between the ER and mitochondria at junctions between the two organelles, where ER membrane and mitochondrial membranes closeted together.
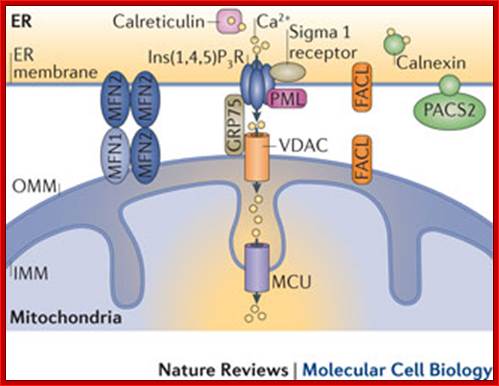
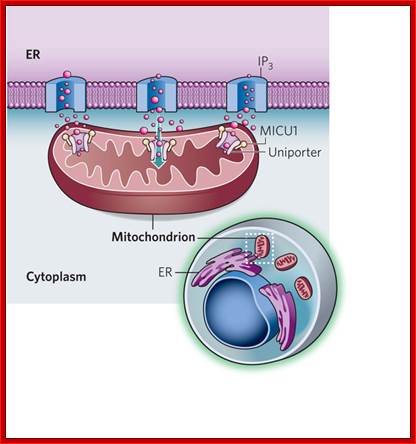
Upper diagram; buildup of the ER-mitochondria junctions; mitochondria as sensors and regulators of calcium signaling; Close interactions between the endoplasmic reticulum (ER) and mitochondria are essential for rapid and sustained Ca2+ uptake by mitochondria. Voltage-dependent anion channels (VDACs), located at the outer mitochondrial membrane (OMM), are responsible for the rapid transfer of Ca2+ from the ER–mitochondria apposition, and their function results in high Ca2+ microdomains in the mitochondria intermembrane space. Accumulation of Ca2+ into the mitochondrial matrix occurs via the mitochondrial Ca2+ uniporter (MCU), which rapidly accumulates Ca2+ across the steep electrochemical gradient. A number of chaperones and regulatory proteins control the formation of the ER–mitochondria junction, the clustering of signalling proteins and their modulation. Mitofusin 2 (MFN2) is involved in both mitochondrial fusion and in ER–mitochondria tethering, by both homotypic interactions and heterotypic interactions with MFN1. Chaperones modulate ER Ca2+ buffering (for example, calreticulin and calnexin) and control the stability or the sorting of signaling proteins. For example, sigma 1 receptor stabilizes inositol-1,4,5-trisphosphate (Ins(1,4,5)P3) receptors (Ins(1,4,5)P3Rs) when ER Ca2+ stores are depleted, thereby ensuring proper Ca2+ fluxes from the ER to the mitochondria. Phosphofurin acidic cluster sorting protein 2 (PACS2) controls the translocation of calnexin from the ER to the plasma membrane and thereby modulates ER Ca2+ buffering and controls ER–mitochondria appositions during apoptosis. Moreover, chaperones affect the activity of ion channels. For example GRP75 (75 kDa glucose-regulated protein), which mediates the interaction of VDAC1 with Ins(1,4,5)P3R, facilitates mitochondrial Ca2+ uptake, and PML (promyelocytic leukaemia) protein, which regulates Ins(1,4,5)P3R-mediated Ca2+ release from the ER, supports mitochondrial Ca2+ uptake and thus has a crucial role during apoptosis. The family of long-chain fatty-acid CoA ligases (FACL) is involved in lipid metabolism and is enriched in mitochondria-associated membranes (MAMs). IMM, inner mitochondrial membrane. Lower diagram: http://www.nature.com; Calcium ions are directly transferred from ER to mitochondrial matrix where the calcium ions are stored for the release with apoptotic signals; Rosario Rizzuto et al, http;//www.nature.com; www.netcategory.net
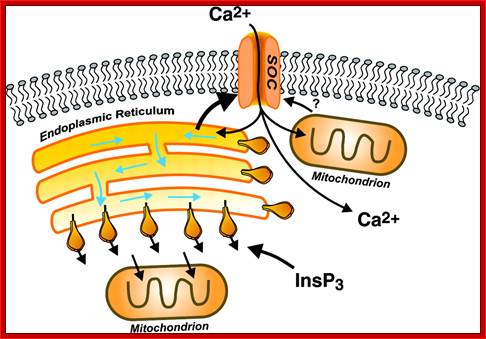
Modes of regulated Ca2+ entry across the plasma membrane. Calcium can enter cells by any of several general classes of channels, including voltage-operated channels (VOC), second messenger-operated channels (SMOC), store-operated channels (SOC), and receptor-operated channels (ROC). VOCs are activated by membrane depolarization, and SMOCs are activated by any of a number of small messenger molecules, the most common being inositol phosphates, cyclic nucleotides, and lipid-derived messengers (diacylglycerol and arachidonic acid and its metabolites). SOCs are activated by depletion of intracellular Ca2+ stores, and ROCs are activated by direct binding of a neurotransmitter or hormone agonist (Ag). In addition, under some conditions, Ca2+ can enter cells via the Na+-Ca2+ exchanger (NCX) operating in reverse mode; http://physrev.physiology.org/
The figure below shows a simple diagram of mitochondrial structure storing Cyt.C.
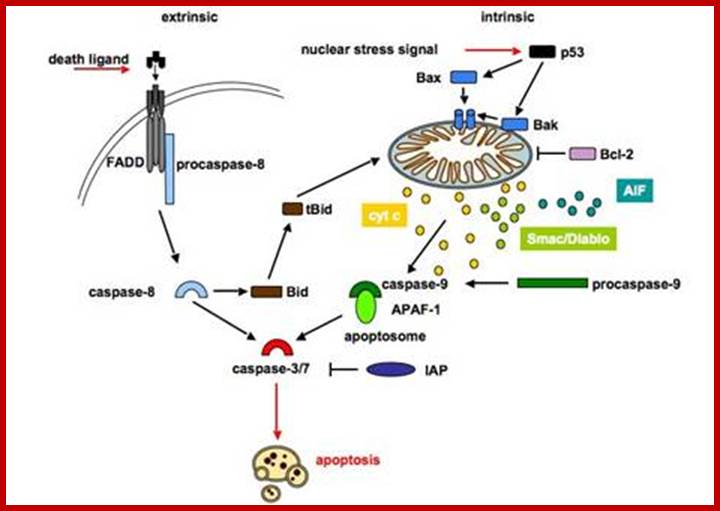
The intrinsic pathway is regulated by proteins of the Bcl-2
family and is therefore referred to as the mitochondrial pathway, because the
mitochondria play a central role (figure 2). Several stimuli can lead to
activation of the mitochondrial cell death pathway such as cytotoxic drugs,
heat shock, ionising, DNA damage and growth factor withdrawal. These stimuli
trigger Bax and Bak activation, which subsequently mediate the permeabilization
of the outer mitochondrial membrane and release of distinct proteins. The mitochondrial (intrinsic) apoptotic signalling pathway
is initiated by cell damaging events, upon which pro-apoptotic members of the
Bcl-2 family are activated and translocate to the mitochondria to neutralize
anti-apoptotic proteins. Permeabilization of the mitochondria causes the
release of cytochrome c. Released cytochrome c associates with Apaf-1 and
procaspase-9 in the presence of dATP to form the apoptosome. Activated
caspase-9 triggers a caspase cascade leading to apoptosis.
The death receptor (extrinsic) pathway is activated when ligands of the
TNF family bind to their receptors on the cell surface. Binding of the ligand
induces trimerization of the receptor and recruitment of the adaptor protein
FADD and caspase-8. Within this complex, caspase-8 is activated and in turn
cleaves and activates casapse-3. The two pathways are mostly independent, but
in type II cells the two pathways can be linked via cleavage of Bid by
caspase-8, caspases-3 or -10. Truncated Bid activates the mitochondrial
apoptotic pathway.
http://edoc.hu-berlin.de
ER pathway:
The endoplasmic reticulum is responsible for the maintenance of the calcium homeostasis and is also the major intracellular calcium storage. The uptake of Ca2+ into the lumen of the ER is managed by energy-dependent SERCA (sarcoplasmic/endoplasmic reticulum Ca2+-ATPase). The release is handled by IP3 (Inositol 1,4,5-Triphosphat (IP3)-regulated receptors or ryanodine (RyR) Ca2+ receptors (Berridge, et al., 2000 ). Furthermore, the ER is the main compartment for protein synthesis, folding, targeting and trafficking. The ER contains numerous chaperone proteins, a high level of calcium and an oxidative environment to carry out these functions efficiently (Rao, et al., 2004 ). Changes in Ca2+ levels or accumulation and aggregation of un- or misfolded proteins lead to ER stress, which is gauged by the ER stress sensors IRE1, PERK and ATF6. To restore normal ER function, the unfolded protein response (UPR) is initiated (Szegezdi, et al., 2006 ). Excessive ER stress forces the unfolded protein response to activate diverse pathways that eventually lead to apoptosis (Ferri and Kroemer, 2001 ). Also, prolonged ER stress is involved in the pathogenesis of some neurodegenerative disorders that feature misfolded proteins (Rao, et al., 2002 ). ER stress can also be elicited by several agents including tunicamycin a specific N-glycosylation inhibitor, Brefeldin A, an inhibitor of the protein transport from ER to Golgi and thapsigargin, which blocks Ca2+ uptake by inhibiting the SERCA (Lee and East, 2001 ). The answer to these stresses is the upregulation of ER chaperons, including the glucose regulated protein GRP78 also referred to as BiP (immunoglobulin heavy chain-binding protein) and the transcription factor CHOP (C/EBP homologous protein). They have both anti-apoptotic features and regulate the ER stress sensors (Lee, 2005 ). To relief the ER stress, GRP78/BiP accelerates protein folding in the ER lumen (Momoi, 2004 ). CHOP sensitizes cells to ER stress by downregulation of Bcl-2 and activation of GADD34 (protein phosphatase 1 (PP1)-interacting protein) and ERO1alpha, an ER oxidase (Li, et al., 2006 ). In summary, these proteins facilitate protein folding and prevent aggregation.
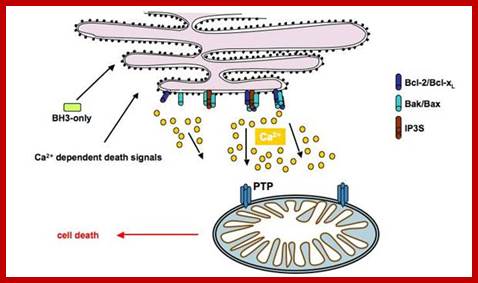
PTPC-dependent apoptotic signaling between ER and mitochondria in response to ER stress. Experimentally induced ER stress (A23187, TG, TN) leads to IP3R-mediated release of ER lumenal Ca2+, mitochondrial accumulation through VDAC and Ca2+-dependent opening of PTPC and L-type calcium channels. The opening of PTPC then promotes PMM, release of apoptogenic factors and subsequent nuclear apoptosis. The proteins involved in the apoptotic pathway triggered by ER stress was determined by pharmacological studies using inhibitors of IP3R (2-APB, 2-AminoethoxydiPhenyl Borate), VDAC (DIDS, dihydro-4,4′ diisothiocyanostilbene-2,2′-disulphonic acid and NADH) and PTPC (CsA ,cyclosporine-A and the-protonophore CCCP, carbonyl cyanide 3-chlorophenylhydrazon). The anti-apoptotic protein Bcl-2 was demonstrated to inhibit PTPC opening in response to ER stress, whereas this latter process was favored by the pro-apoptotic protein Bax; mitochondria stores Ca2+ in its matrix.; Apoptotic signals targeting the ER by the release of cytochrome C, may induce Ca2+ release to regulate mitochondrial activation by opening its permeability transition pore (PTP).; http://www.bioscieence.orghttp://edoc.hu-berlin.de
Crosstalk between ER and mitochondria;
The mechanisms and the part of crosstalk between the mitochondria and the endoplasmic reticulum are not entirely illuminated. But it seems that cytochrome c induced apoptosis activated by ER - mitochondria crosstalk is important for ER stress mediated cell death (Momoi, 2004 ). Moreover, the main signal in ER-mitochondria crosstalk is thought to be calcium. After activation of the death receptor pathway and consequent activation of caspase-8, BAP31 (Bcl-2-associated protein 31) is cleaved to a p20 fragment. BAP31 is an integral ER membrane protein that seems to be a mediator of crosstalk between the two organelles and it has pro-apoptotic capacities. It is cleaved and activated by a unique isoform of caspase-8 (Breckenridge, et al., 2002 ). The p20 cleavage product of BAP31 causes the release of Ca2+ from the ER. Liberated Ca2+ is taken up by the mitochondria inducing the recruitment of Drp1. Drp1 mediates the scission of the outer mitochondrial membrane, resulting in dramatic fragmentation and fission of the mitochondrial network and cytochrome c release (Breckenridge, et al., 2003 ). Calcium signals from the ER regulate the opening of the permeability transition pore. Absorbed Ca2+ in the matrix causes at a certain level the opening of the mitochondrial transion pore, which leads to loss of the mitochondrial membrane potential and hence to the release of cytochrome c and apoptotic factors. This process is followed by translocation of cytochrome c to the ER, where it interacts with IP3 receptors to induce a positive feedback loop;http://edoc.hu-berlin.de
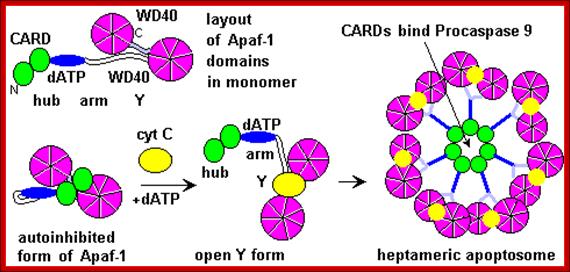
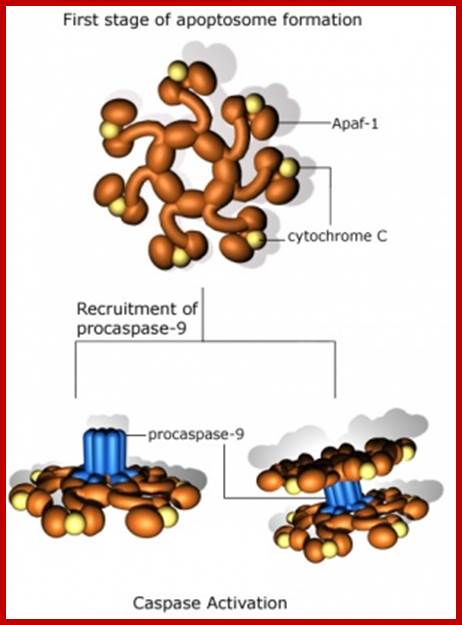
The figure above and the figure below shows the released Cyt.C binding to Apaf-1 at the domain called CARD, and further activated by the binding of ATP. This Y-complex binds to procaspase-9 and activates to cleave it and form a heptameric complex called Apoptosome, which in turn activates procaspase-3 that leads to devastating effect on cellular components and cell death.
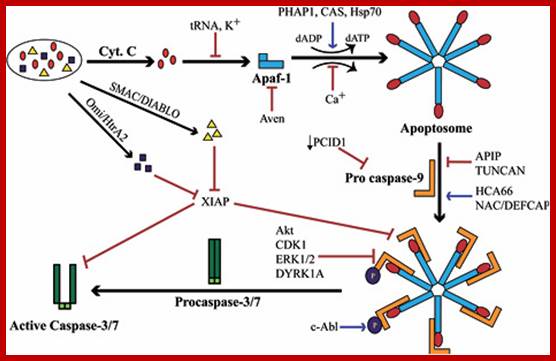
Post-MOMP regulation of apoptosis. Following release from the mitochondria, cytochrome c binds Apaf-1 and triggers the formation of a heptameric wheel-like complex, the apoptosome, which recruits and activates caspase-9. Proteins like Aven, physiological levels of nucleotides like tRNA and ATP, as well as intracellular K+ can all inhibit this process by directly inhibiting the interaction between Apaf-1 and cytochrome c. Formation of the apoptosome also requires nucleotide exchange on Apaf-1, a process stimulated by a combination of three proteins: PHAP1, Hsp70, and CAS, and inhibited by intracellular Ca2+. Recruitment of procaspase-9 to the apoptosome is antagonized by APIP and TUNCAN, and stimulated by HCA66 and NAC/DEFCAP. Furthermore, downregulation of PCID1 causes concomitant decrease in procaspase-9 levels. Direct phosphorylation at Thr125 by Akt, CDK-cyclin B1, ERK1/2, and DYRK1A inhibits caspase-9 activity through unclear mechanisms. Conversely, phosphorylation at Tyr153 stimulates activation. XIAP mediated inhibition of caspase-9 and caspase-3 activity occurs through distinct mechanisms, and in both cases, this repression is relieved by SMAC/DIABLO and Omi/HtrA2, which are also released from the mitochondria following MOMP; http://exp-oncology.com.ua
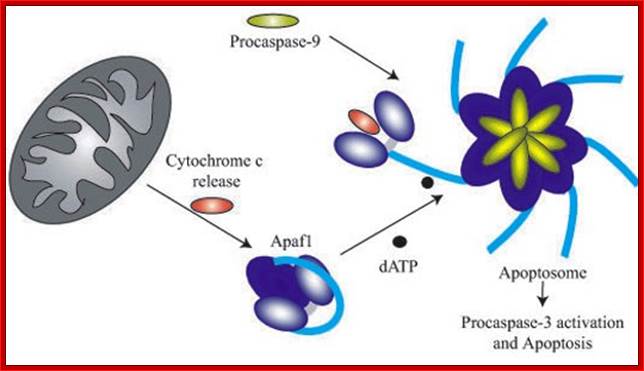
The conformational changes in APAF1 lead to Apoptosome formation and activation of apoptosis; http://atlasgeneticsoncology.org/
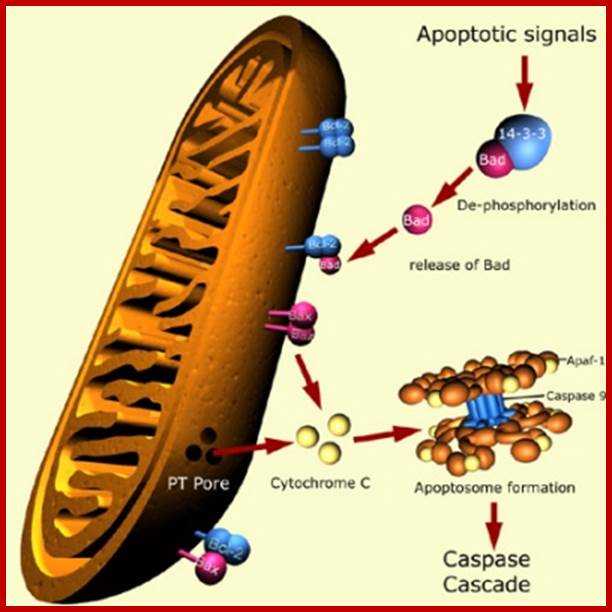
First they cluster as trimers and more clusters develop. The activated cytosolic domain now binds with adaptor proteins such as FADD in the case of Fas 1-R and TRADD and FADD in the case of TNF a1-R. All are in active state. At this juncture cellular precursor Casp-8 interacts with the adaptor proteins through its own death domains, this complex is called death inducing signal complex (DISC), which in turn activates, Casp-8. The activated Casp-8 performs auto cleavage of its own structure into large and small subunits and they bind to each other as dimeric proteins.
Apoptosis- mitochondrial pathway: https://cellbiology.med.unsw.edu.au/
Apoptosome formation; https://cellbiology.med.unsw.edu.au/
Activated proteins can directly act on casp-3 and activates the death process. The second route is to act on Bid to activate mitochondrial mediated process.
This activated casp-8 (as dimeric protein) now acts on Bid protein and cleaves into N-end and C-end subunits. The c-end subunit translocates into outer mitochondrial membrane and interacts with Bax proteins. Found. In the membrane the Bax proteins are associated with another class of proteins called Bcl2. If the Bcl2 exist as homodimers, interaction of Bid fails to release Cyt-C from the periplasmic space of Mitochondria. But on the contrary if the Bax is associated with Bcl2 as heterodimers, they facilitate the release of Cyt-C into cytosol. The concentration of Bcl2 and Bax is very important. If the Bax proteins are in homodimeric state, Cyt-c will be released. If the Bcl2 are in homodimeric state, Cyt-c is not released, but if the Bax and Bcl2 are in heterodimeric state, the Bid interaction induces them to release Cyt-C. The Bcl2 protein when expressed in higher amounts, it prevents apoptosis and induces cell proliferation.
The released Cyt-C interacts with pro-casp-9 and activates it through another protein called ApaF1. First Cyt-c binds to ApaF1 and this dimer binds to ATP. The activated dimers interact with pro-Casp-9 and cleave it to active casp-9. This complex of Cyt-c, ApaF1 and casp-9 is called Apoptosome. Such trimers can interact with one another and form a ring of huge Apoptosome complex.
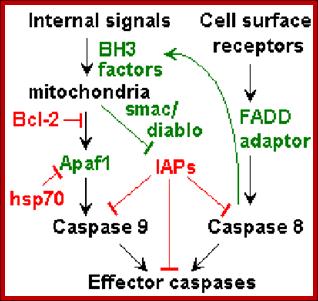
The Apoptosome acts on pro-casp-3 (ced3). The casp-9 also activates casp-6 and 7. The activated casp-3 activates some downstream proteins such as Caspase activated DNAase (CAD). This DNAase is found in cytoplasm, but an inhibitor called ICAD binds it. Caspase-3 degrades the ICAD and releases the DNAase in active form, which enters into the nucleus and cleaves the DNA. Though there are Inhibitors of apoptosis (IAPs) within the cell, they are activated casps. Diablo inactivates IAPs. Mitochondrial also releases endonuclease G that enters the nucleus and cleaves DNA into 180 bp long fragments. Ced-3 virtually dismembers all cellular protein especially of microtubules and other cytoskeleton proteins. This act of enzyme causes blebbing and destruction of cells.
Signal from inside:
When the cellular DNA is damaged because of errors in replication or damaged by irradiations, or due to the loss of protection of Telomeric DNA, these events lead to fragmentation of DNA with 3’hanging ends. Such fragments are recognized by p53 proteins through Rad9; and the p53 gets activated via phosphorylation by Akt members. P53 proteins are considered as tumor suppressors. Binding of p53 to DNA ends activates the protein, the activated tetramers activate the transcription of certain genes, and one such is the gene for p21 and p27 proteins. The p21 and p27 proteins inhibit the activity of Cdk-Cyclin complexes and hold the cell at G1-S transitional stage. The activated p53, if finds the DNA damage is severe, the p53 by its N-terminal domain interacts with mitochondrial membranes and promotes the release of Cyt-C, which activates the whole process of cell destruction. It is for this reason the N-terminal region of p53 is called ‘apoptotic domain’. The actual apoptotic domain lies within the first 63 amino acid residues from the N-end; especially residues 53 and 54 are critical for both apoptotic and transcriptional activation.
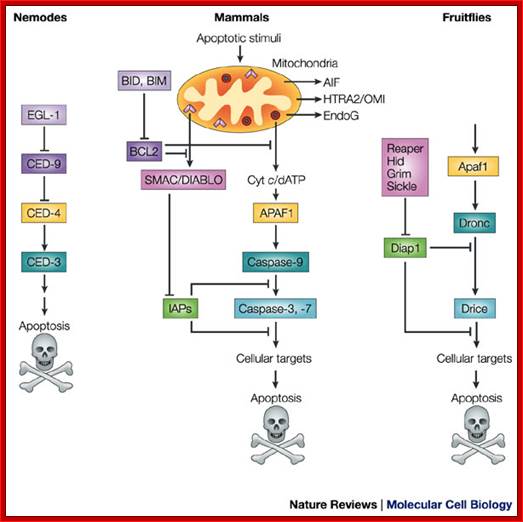
A conserved apoptotic pathway in nematodes, mammals and fruitflies. Molecular mechanisms of caspase regulation during apoptosis; Stefan J. Riedl and Yigong Shi; http://www.nature.com/
Functional homologues of caspases and caspase regulators across species are indicated by the same colour. Caspase-9 in mammals and Dronc in the fruitflyDrosophila melanogaster are initiator caspases, whereas caspase-3 and -7 in mammals and Drice in fruitflies belong to the class of effector caspases. CED-3 (cell-death abnormality-3) in the nematode worm Caenorhabditis elegans functions both as an initiator and effector caspase. The inhibitor of apoptosis (IAP) proteins suppress apoptosis by negatively regulating the caspases, whereas SMAC (second mitochondria-derived activator of caspases)/DIABLO (direct IAP-binding protein with low pI) in mammals and the RHG proteins Reaper, Hid, Grim and Sickle in fruitflies can remove the IAP-mediated negative regulation of caspases. AIF, apoptosis-inducing factor; APAF1, apoptotic-protease-activating factor-1; Cyt c, cytochrome c; EndoG, endonuclease G; HTRA2, high-temperature-requirement protein A2. Stefan J. Riedl and Yigong Shi.
Stefan J. Riedl and Yigong Shi
Other pathways:
1. Fas can also be activated to execute apoptotic pathway by using JNK kinase, whose substrate is C-Jun a transcription factor. This pathway activates certain set of proteases. When the signals are impinged upon the Fas-R, the activated receptor proteins interact at cytosolic side with DaXX. The TNF- receptor can also be activating JNK pathway by another distinct adaptor proteins, however the stress related activation of JNK is independent of Fas pathway, which is not inhibited by Bcl2. The intermediate reaction in JNK pathway starts from Fas-R/TNF-R. The activated Fas-R/TNF-R binds to DaXX at cytosolic surface. They in turn interact with JNK system, which then phosphorylates C-Jun. The phosphorylated c-Jun. is involved in production of Caspases that leads to apoptosis.
2. In the absence of death inducing signals, where cells are destined to die, cells do die by their own self-suicidal mechanism. Do human beings have that mechanism? When the cell is aged and the cell death receptor not activated or in the absence of Trophic factors, a cytosolic protein called Bad (Bcl-xl/Bcl2 associated X-protein), associates Bcl2-BclX1 proteins found at the outer membrane of mitochondria and activate the transport system to release Cyt.C., that activates a cascade of reaction leading to the death of the cells.
The fascinating aspect of this phenomenon is, in presence of a mitogenic factor, when the ligand binds, the cytosolic domain activates Phosphotidyl inositol-3 kinase PI-3. The activated PI-3 activates AKT kinase, which is another protein kinases; it phosphorylates Bad protein. The phosphorylated Bad proteins are sequestered by an un-usual protein called 14.3.3. this blocks the release of Cyt.C. thus blocks cell death by suicide.
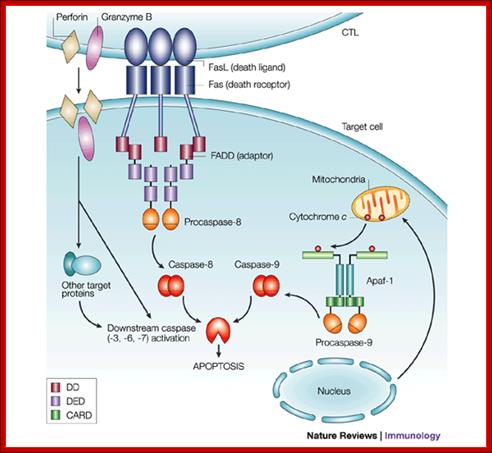
T-Cell mediated cytotoxic pathway:
In the case of cytotoxic mediated cell death, specific cellular proteins at their surface mark the cells. The activated cytotoxic T-lymphocytes bind to such surface proteins and kill their target cells by releasing Granzymes in the form of granules. Perforin creates holes in the receiver cells, thus allow all the enzymes to enter into the target cell. Granzyme-B induces many features similar to apoptosis including DNA fragmentation. It activates Caspase-3, which is very essential for apoptosis.
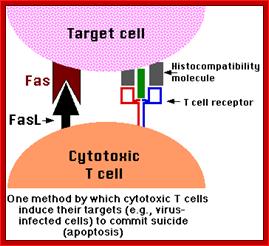
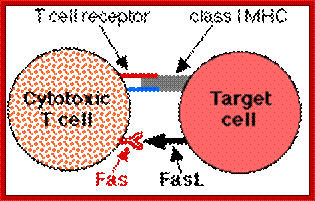
Apoptosis; http://users.rcn.com/
By using this technology, it is possible to destroy tumor cells by a process called cell-mediated therapy. Tumors contain their own lymphocytes called Tumor infiltrated lymphocytes called TILs. Such TILs can be recovered and activated by specific interleukins. The activated TILs become killer cells. When such activated TILs are injected into the patient TILs reach their specific tumors and by binding they release Granzymes and kill the targets cells, thus it is possible to remove tumors.
Apoptosis vs Carcinogenesis:
Genetic studies on C. elegans has revealed programmed death of those 130 cells among 1090 cells is due to certain genes. Some of them are pro-apoptotic and some of them are anti-apoptotic. Similar genes and more have been identified in human systems. Gene knockout experiments using cultured transgenic cells, the role of each of the genes has been elucidated to some extent. In C.elegans the genes CED3 and CED4 leads to cellular death and CED-9 prevents cell death. The counterparts in human system are Bcl-2, Apaf-1, Casp9 and Casp3; the first one Bcl2 is a suppressor of apoptosis and the remaining three are pro-apoptosis factors.
C.elegans: Ced-4-àCed-3-àCell death, this path way is blocked by Ced-9.
Human: Apaf-1-à Casp-9 -à Casp3 -àcell death; this pathway is blocked by Bcl-2.
Expression of CED4 in kidney cells leads to rapid apoptosis. This can be blocked by the co-expression of Bcl2 or CED9. CED4 binds to CED3 and activates protease activity that leads to the degradation of cellular proteins, hence cell death. Bcl2 and CED9 are homologous proteins found on the outer membrane of mitochondria, endothelial membranes and outer nuclear membranes. These proteins have a single transmembrane domain
The key cellular component responsible for destruction of cellular proteins is Caspase. It is a cysteine-protease when activated, targets proteins importantly cellular microtubules and nuclear lamins and degrade them by cutting the peptide bond towards the carboxyl side of the Aspartate amino acid; that is the reason why nucleus and cells collapse and crumble. In C. elegans the most effective molecule is CED3. In mammals they have multiple Caspase. CED9 binds to CED4 and localizes from the cytosol to cellular membranes. So CED’s apoptotic function is suppressed but CED4 binds to CED3 and activates protease activity.
Another pro-apoptotic factor is Bax, which is found associated or complexed with Bcl2, but it’s over expression causes apoptosis. Sequence of Bax protein is similar to that of CED9 and Bcl2, but over expressing Bax induces death of cells. All these proteins have single transmembrane domains and enable oligomerization.
Bcl2 belongs to a family of proteins involved in distribution of Cytochrome-C, which in normal conditions found in the space between outer and inner mitochondrial membranes; actually, this is a marker protein. Bcl2 blocks the release of this protein, but Bax counteracts the Bcl2 and promotes the release of Cyt.C into cytoplasm. The released Cyt.C binds to apoptosis activation factor-ApaF-1 (CED-4) and activates Caspase cascade.
The release of Cyt.C from the mitochondrial membranes into the cytosol is
due to the influx of ions from the cytosol; which is due to the binding of homo-
dimers of Bax but not Bcl2/Bax heterodimers to mitochondrial membranes. Bcl-xl is another anti-apoptotic protein. When this gene is knocked out of mice there is a massive cell death in spinal cord and brain regions, but knock out of Bax leads to marked increased neuronal tissues.
Trophic factors work independent of protein synthesis. Binding of Trophic factors to their respective receptors induce signal transduction and down stream signaling process where certain proteins are post translationally modified. The Bad protein in it non-phosphorylated form associates with Bcl2/Bcl-xl at mitochondrial membranes. Binding of Bad (-P) to Bcl2/Bcl-xl inhibits anti-apoptotic function of Bcl2/Bcl-xl. But phosphorylated Bad (+P) fail to bind to Bcl2/Bcl-xl, for phosphorylation leads to sequestering of bcl2/Bcl-xl by phosphoserine binding proteins in cytosol.
Certain growth factors activate PI-3 kinase, which in turn can activate downstream kinase such as Akt. This kinase phosphorylates Bad at specific sites; this is known to inhibit pro-apoptotic activities. Constitutively active Akts can rescue cultured cells that are deprived of neutrophin from death.
As in the case of cell proliferation, mitogen factors stimulate cell proliferation; if it is not controlled it leads to cancer. Similarly certain damage to genetic material in the cells, which is beyond repair system to normality, certain molecular sensors with in the cells, sensing the severity of damage; trigger the process of cell death, called Apoptosis.
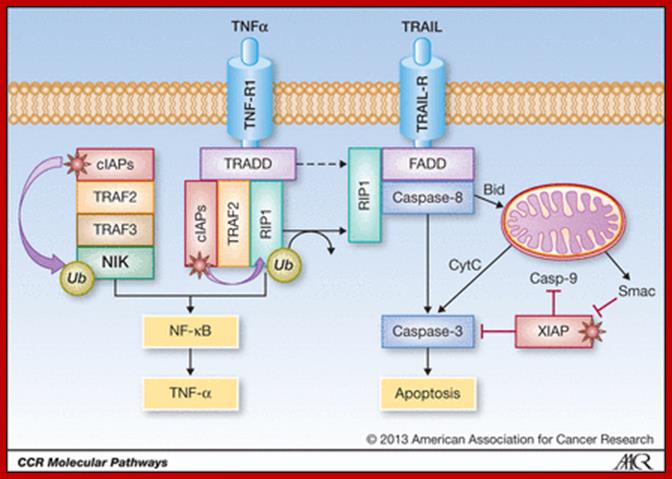
Regulation of apoptosis and NF-κB signaling by IAP proteins and their antagonists. XIAP negatively regulates both the extrinsic and intrinsic apoptosis pathways by inhibiting caspases-3 and -9. The extrinsic (death receptor) pathway is triggered upon ligation of death receptors such as TRAIL receptors (TRAIL-R) by their ligands such as TRAIL, leading to activation of caspases-8 and -3. The intrinsic (mitochondrial) pathway is engaged by the release of mitochondrial proteins such as cytochrome c (CytC) or Smac into the cytosol, which promotes caspase-9 and -3 activation. Neutralization of XIAP-mediated caspase inhibition by IAP antagonists promotes caspase-dependent apoptosis. cIAP proteins (cIAP) control activation of canonical and noncanonical NF-κB pathways. cIAP proteins promote canonical NF-κB activation via nondegradative ubiquitination of RIP1, whereas they inhibit noncanonical NF-κB signaling via ubiquitination of NIK and its degradation via the proteasome. IAP antagonists stimulate the E3 ubiquitin ligase activity of cIAP proteins, thereby promoting their autoubiquitination and proteasomal degradation. In turn, NIK is stabilized and activates noncanonical NF-κB signaling. Induction of NF-κB target genes such as TNF-α can then, in an autocrine/paracrine manner, trigger TNFR1-mediated caspase-8 activation and apoptosis via a RIP1/FADD/caspase-8 cytosolic complex. IAP antagonists suppress activation of the canonical NF-κB pathway by depleting cIAP proteins. Initially, however, they may stimulate canonical NF-κB signaling by increasing the E3 ubiquitin ligase activity of cIAP proteins, which leads to RIP1 ubiquitination and NF-κB activation. Stars designate IAP antagonists and their targets. Please see the text for more details. Simone Fulda http://clincancerres.aacrjournals.org/
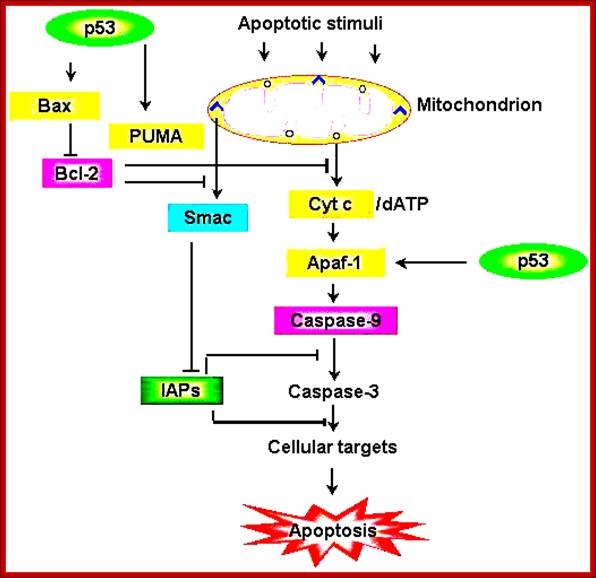
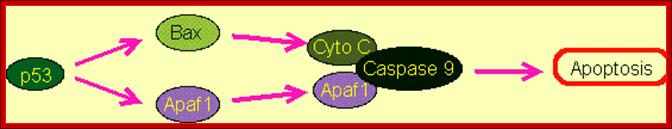
Schematic representation of the p53-dependent
apoptotic pathways by transcriptional
activation of BAX, PUMA and APAF-1.; https://webhome.weizmann.ac.il/
Some Acronyms n Expansions:
APAF1 = Apoptotic protease activating factor.
Apoptosome: Apf1-Cyt-C, dATP-Pro Caspase9-ced 4 complex.
BAD=BCL2 antagonist of cell death.
BAX=BCL2 associated X protein.
BCL2= B-cell Cell lymphoma 2.
Bcl X = B cell cell lymphoma factor that inhibits cell death.
BID= BH3 interacting domain death agonist.
Ced = Cell death genes, 1--XXX
DAXX= death associated protein 6.
DR = Death receptors.
FADD= Fas (TNFRSF^) associated death domain.
FAS = CD95, and Apo 1.
FASL = Fas ligand
TRADD= TNFRSF1A-associated death domain.
Ces= cell specificity.
ICE = Interleukin converting enzyme Caspase 1.
MICE = Min ICE.
PFP = Pore forming proteins.
PI = Propidium iodide, a DNA dye for excluding dead cells.
TRADD = TNF receptor associated death domain.
TRAIL = Tumor necrosis factor related apoptosis inducing ligand.
RAIL = Receptor of TRAIL.
TRAMP = TNF receptor apoptosis mediated protein.
Cell Proliferation and Apoptosis:
There is a relationship between tumor formation and apoptosis. P53 activates several numbers of genes involved in cell cycle regulation. ADV’s E1B, a 19KD protein blocks P53 ‘s transcriptional activation. Bcl2 is an Oncogene identified; its activation is due to few genes identified in tumors. It actually blocks the pathway of apoptosis. So Apoptosis inhibits tumorigenesis and Bcl2 prevents apoptosis. P53 has pleotropic phenotypic effects; it triggers growth arrest and induces apoptosis. RB and P53 are activated in multiple routes. One locus that influences both RB and P53 is INK4A-ARF. Its transcript is alternately spiced as P16NK4A in RB and p19ARF in P53.
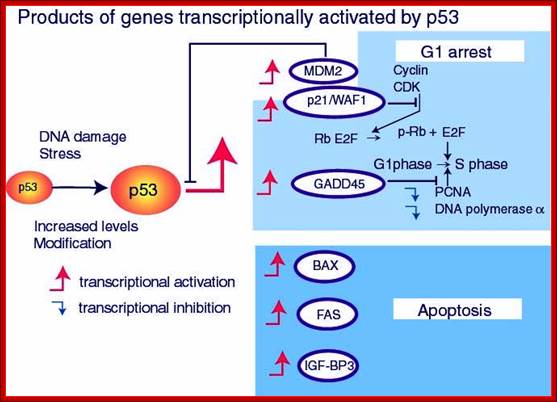
www.netcategory.net
In human cancers deletion of INK4A-ARF is common which eliminates both P16INK4A and P19ARF, thus cell loses the ability to block tumor formation. P16INK4A inhibits cdk4/6 kinase, so it prevents the kinase from phosphorylation RB. In the absence of it cell cycle progression is halted in its track. The activity of INK4A is often disabled because of several point mutations in human tumors. P19ARF antagonizes Mdm2, which leads to stabilization of p53. Thus, p19ARF acts as a tumor suppressor by inhibiting the inhibitor of P53. P19 may promote the degradation of Mdm2 or directly blocks interaction with p53. P19ARF arrests the cell cycle in p53 dependent manner. Loss of PARF and p53 has the same effect.
C-Myc and E1 (ADV) act via p19ARF to activate p53 dependent activation. P53 is often phosphorylated at serine residues (S6, S9, S37etc). P53 is also acetylated at different positions. Ionizing radiations activate the kinase ATM, which phosphorylates S13 and S33 and S376 and L382. Such modifications may affect the stability, oligomerization, DNA binding, binding to other proteins. So p53 acts as a molecular sensor.