RNA Processing and-Editing of mRNA Transcripts:
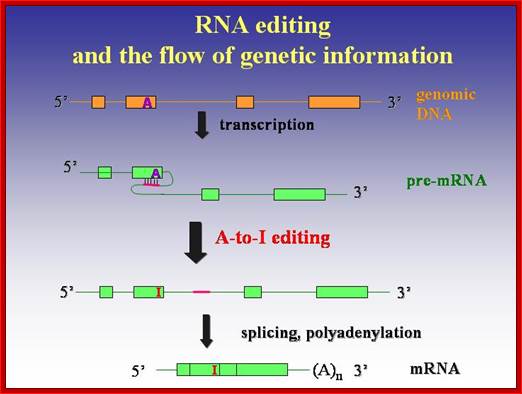
It is an accepted fact that DNA directs to produce RNAs and RNAs execute their functions for what they have been produced, ‘what you have & what U get’, whatever it can be. Here the master and the executive are complementary to each other in most of the cases. This is a general rule, but in certain exceptions, the mRNA produced; a copy of the master, for a functional protein, is different from what is required. In this context the mRNA transcribed cannot be translated to a product that is required and desired by specific cell type. Many such genes are transcribed with certain sequences missing or sequences not required or some sequence in excess, so they have to be substituted, removed or modified. But in many cases the same mRNA produced by the same gene in different tissue types is modified by single bases of a specific nucleotide to the needs of the cell types. However, the molecular mechanisms have been devised to correct the wrong or extra or missing alphabets of the codons, either by removing individual nucleotides or adding individual nucleotides. Gene economy comes into play in this process. The said mechanism is called ‘Editing’, similar to editing of the script that goes to final printing; often this is also called as ‘Proof Reading’. One potential function for this widespread editing of introns in pre-mRNAs is to counteract the deleterious effects of double-stranded RNA, including activation of the PKR kinase and the interferon response. In addition, RNA-editing of introns may play a critical role in evolution (see Herbert, 1996). Human genomic transcripts are edited at perhaps one million or more sites, nearly 90% of the transcripts are edited.
RNA editing:
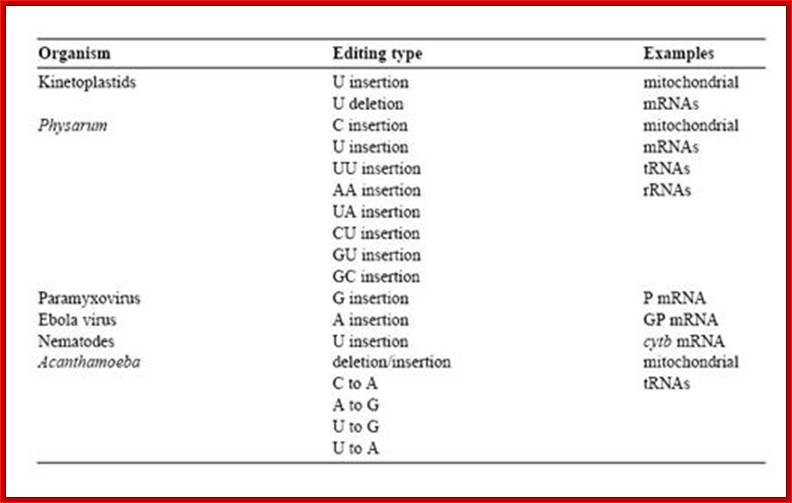
RNA editing is the co- or post- transcriptional modification of primary sequence of RNA that encoded in the genome through nucleotide deletion, insertion, or base modification mechanisms. RNA editing is mainly classified into two classes. One is substitution editing (replacement of individual nucleotides), and the other is insertion/deletion editing (insertion or deletion of nucleotides in the RNA). Different forms of RNA editing are listed in Table 1.1 (substitution editing) and Table 1.2 (insertion/deletion editing). RNA editing is quite widespread occurring in mammals, viruses, marsupials, plants, flies, frogs, worms, squid, fungi, slime molds, dinoflagellates, protozoa, and other unicellular eukaryotes. It should be noted that the list most likely represents only the tip of the iceberg.
Editing of Apolipo-protein mRNA:
Apolipoprotein is produced in different forms in tissue specific manner. The Apolipo protein synthesized in liver has a molecular mass of ~502 KD. Full-length apolipoprotein B100 are metabolized in the blood stream to low-density lipoprotein (LDL) particles, whose elevated levels increase the risk of atherosclerosis. Statins and bile-acid sequestrants are effective LDL-lowering therapies for many patients; The mRNA is larger than 16kb; Mol.Pharmocol 2002.
Editing of Apolipoprotein-B mRNA; http://users.rcn.com/jkimball.ma.ultranet
Table 1. 1 Editing distribution: substitution editing.
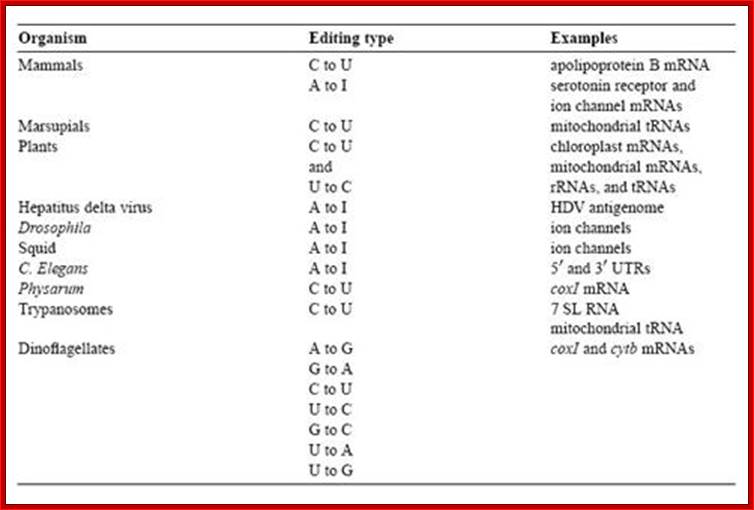
Table 1. 2 Editing distribution: insertion/deletion editing.
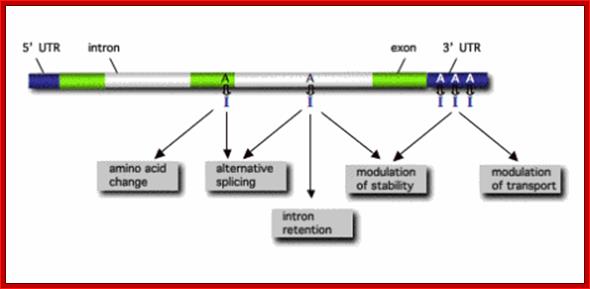
RNA editing in the mammalian brain; One area that is known to require extensive protein diversity is the brain. It is well known that pre-mRNA processing such as alternative splicing is widespread and highly regulated in the nervous system. Another way to alter the mRNA is by RNA editing. Adenosine to inosine (A-to-I) deamination is the most common type of RNA editing found in mammals and it is catalyzed by adenosine deaminases that act on RNA (ADARs). There are two enzymes found to be active in adenosine deamination, ADAR1 and ADAR2. These enzymes convert A-to-I within double-stranded or highly structured RNA. Since inosine is recognized as guanosine by the cellular machinery, A-to-I editing has the potential to change the code for translation. Site-selective A-to-I editing is a mechanism used to fine-tune the transcriptome and increase the variety of expressed protein isoforms, mainly found in the central nervous system (CNS). Besides amino acid changes, editing has been found to be able to influence the outcome of splicing as well as other post-transcriptional events like 3’ end processing, stability, and transport (Figure 1). Thus, RNA editing by A-to-I modification has the power to affect the proteome in many different ways. http://www.su.se/
Editing of Apolipo protein mRNA:
Apolipoprotein is produced in different forms in tissue specific manner. The Apolipo protein synthesized in liver has a molecular mass of ~502 KD. Full-length apolipoprotein B100 are metabolized in the blood stream to low-density lipoprotein (LDL) particles, whose elevated levels increase the risk of atherosclerosis. Statins and bile-acid sequestrants are effective LDL-lowering therapies for many patients; The mRNA is larger than 16kb; Mo. Pharmocol 2002.
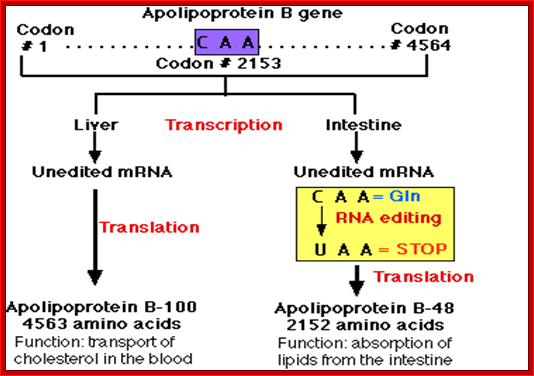
The human APOB gene; Apolipoprotein B-100 (apoB-100; Humans have a single locus encoding the APOB gene
- It contains 29 exons (separated by 28 introns).
- The exons contain a total of 4564 codons.
- Codon 2153 is CAA, which is a codon for the amino acid Glutamine (Gln).
- The gene is expressed in cells of both the liver and the intestine.
- In both locations, transcription produces a pre-messenger RNA that must be spliced to produce the mRNA to be translated into protein,
- In the Liver: Here the process occurs normally producing apolipoprotein B-100 — a protein containing 4,563 amino acids — that is essential for the transport of cholesterol and other lipids in the blood.
- In the Intestine.
- In the cells of the intestine, an additional step of pre-mRNA processing occurs; the chemical modification of the C nucleotide in Codon 2153 (CAA) into a U occurs.
- This RNA editing changes the codon encoding the amino acid glutamine (Gln) to a STOP codon (UAA).
o The modification is catalyzed by the enzyme cytidine deaminase that recognizes the sequence of the RNA at that site in the molecule and catalyzes the deamination of C thus forming U.
- Translation of the mRNA stops at codon #2153 forming Apo lipoprotein B-48 — a protein containing 2152 amino acids — that aids in the absorption of dietary lipids from the contents of the intestine.
At its N- terminal, it has a lipid-binding domain, and the C-terminal has a LDL receptor binding domain. It is through the interaction between Apolipo protein and the receptor, lipids are transported into cells.
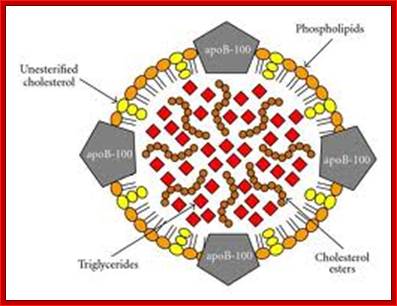
Antigen Induced Immunomodulation--Schematic representation of the low-density lipoprotein particle (LDL). The LDL particle has a size of approximately 21–24 nm and is the main transporter of unesterified cholesterol, cholesterol esters, and triglycerides in the blood. It contains an outer layer composed of phospholipids and unesterified cholesterol in which a single protein is embedded, the apolipoprotein B-100 (apoB-100). These components are more susceptible to oxidation by free radicals in the sub endothelial space during inflammation. They are also targets for the recognition of the LDL by scavenger receptors, proteoglycans, and low-density lipoprotein receptor (LDLr). The core of the particle contains primarily cholesterol esters and triglycerides. In atherogenesis, a large number of IgM antibodies are created in response to oxidative stress-modified phospholipids, whereas IgG antibodies and T-cell clones are generated against apoB-100.; http://www.hindawi.com/
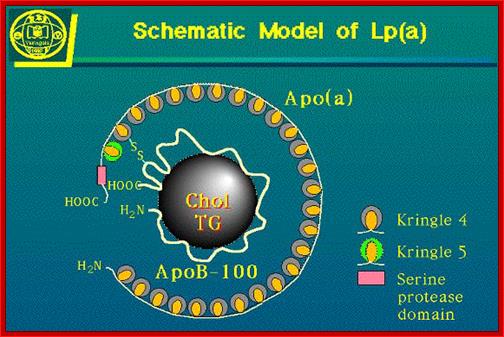
Schematic Model of Lp (a)
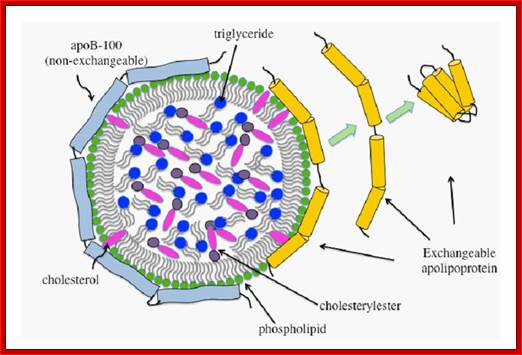
Schematic representation of a generic lipoprotein particle. Lipoproteins have a spherical geometry with a monolayer of amphipathic lipids and proteins encircling a core of neutral lipids. ApoB-100 (pale blue) is a single large polypeptide and is the non-exchangeable component of lipoproteins such as VLDL and LDL. The molecular mass of exchangeable lipoproteins (yellow cylinders) varies from 8-50 kDa. The exchangeable apolipoproteins have the ability to exist in lipid-free and lipid-bound states. Vikram Jairam, Koji Uchida and Vasanthy Narayanaswami http://www.intechopen.com/
The same protein however, is produced in intestine and it does not require interaction with cellular LDL receptors and so the protein is lacking in receptor binding domain. The Apolipo protein is produced in different forms in different tissues for the proteins produced in tissue specific manner have different functions.
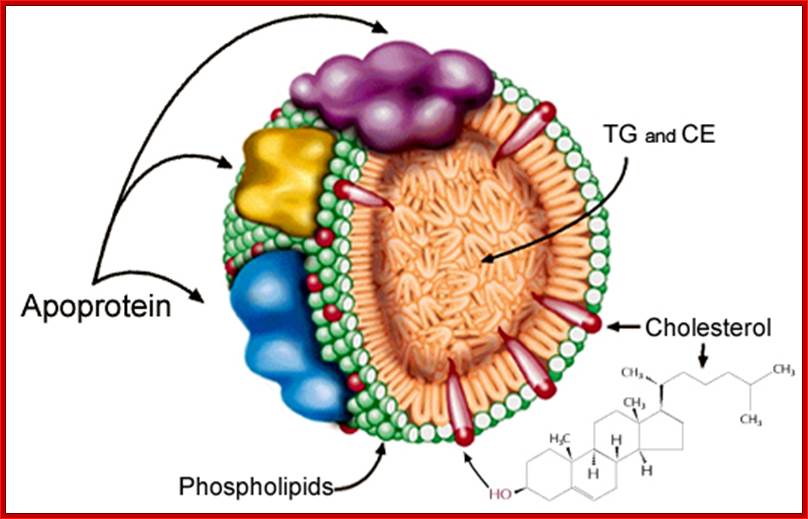
Schematic representation of human and insect lipoprotein; there are two types of Apo lipoproteins: 1.the non-exchangeable protein, apolipoprotein B or ApoB, present in all lipoproteins except HDL 2. The exchangeable proteins (Apo A, C,) which are absent in LDL; in insects they are called-Lipophorins. http://intranet.tdmu.edu.ua/
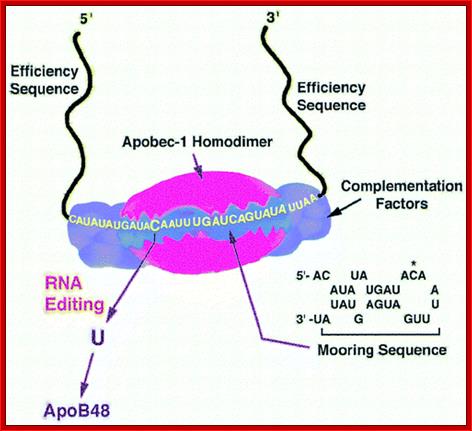
Apolipoprotein (apo)B circulates in two distinct forms, apoB100 and apoB48. Human liver secretes apoB100, the product of a large mRNA encoding 4564 residues. The small intestine of all mammals secretes apoB48, which arises following C-to-U deamination of a single cytidine base in the nuclear apoB transcript, introducing a translational stop codon. This process, referred to as apoB RNA editing, operates through a multicomponent enzyme complex that contains a single catalytic subunit, apobec-1, in addition to other protein factors that have yet to be cloned. ApoB RNA editing also exhibits stringent cis-acting requirements that include both structural and sequence-specific elements—specifically efficiency elements that flank the minimal cassette, an AU-rich RNA context, and an 11-nucleotide mooring sequence—located in proximity to a suitably positioned (usually upstream) cytidine. C-to-U RNA editing may become unconstrained under circumstances where apobec-1 is overexpressed, in which case multiple cytidines in apoB RNA, as well as in other transcripts, undergo C-to-U editing. ApoB RNA editing is eliminated following targeting of apobec-1, establishing that there is no genetic redundancy in this function. Under physiological circumstances, apoB RNA editing exhibits developmental, hormonal, and nutritional regulation, in some cases related to transcriptional regulation of apobec-1 mRNA. ApoB and the microsomal triglyceride transfer protein (MTP) are essential for the assembly and secretion of apoB-containing lipoproteins. MTP functions by transferring lipid to apoB during its translation and by transporting triglycerides into the endoplasmic reticulum to form apoB-free lipid droplets. These droplets fuse with nascent apoB-containing particles to form mature, very low-density lipoproteins or chylomicrons. In cultured hepatic cells, lipid availability dictates the rate of apoB production. Unlipidated or underlipidated forms of apoB are subjected to presecretory degradation, a process mediated by retrograde transport from the lumen of the endoplasmic reticulum to the cytosol, coupled with multi-ubiquitination and proteasomal degradation. Although control of lipid secretion in vivo is primarily achieved at the level of lipoprotein particle size, regulation of apoB production by presecretory degradation may be relevant in some dyslipidemic states. Apo lipoprotein B: mRNA Editing, Lipoprotein Assembly, and Pre secretory Degradation; Annual Review of Nutrition.
· The LDL receptor gene is same in all tissues and the mRNA produced after splicing, is also same in all tissues.
· Even though all genes, found in all cells and tissues in a given organism, show heterogeneity 45% or more.
· The LDL receptor mRNA is nearly 13800 to 13900 nucleotides long. The pre-mRNA has 29 Exons. It codes for 4564 amino acids. In liver the cis-spliced but unedited mRNA is translated to produce the regular protein, this protein is called Apolipo protein B-100.
· In intestine cells, the codon CAA (Gln) found in 26th Exon at 2153 th codon, is changed into UAA, by an intestinal cellular enzyme, which removes amino group from C and adds an oxygen in its place to form U; the process is called deamination.
· The UAA happens to be a translational terminator, and the protein produced is truncated at this codon and it is 2152 amino acids long, hence this protein is called Apolipo protein B-48. This change is made possible because the required enzyme is produced in intestinal cells. This enzyme works on ds-loop region containing CAA and as the recognition site and converts cytosine to uracil by deamination and oxidation, to generate uracil.
· The enzyme is cytosine deaminase, and it is highly specific to the nucleotide. Hence, intestinal cells are endowed with factors to change the codon as well as the character of the protein.
)
ApoB
pre-mRNA is encoded with numerous exons that have to be spliced in order to
create a mature mRNA. Editing occurs at position 6666 (CAA) within exon 26.
This introduces a premature stop codon up stream of the native stop codon
within the terminal exon (UAA). For reasons that are not totally clear but
appear to involve ACF binding to the mooring sequence, edited apoB mRNA is not
subjected to nonsense mediated decay (NMD).
ApoB mRNA editing was first suggested to be a nuclear process from the analysis of the proportion of edited apoB mRNA among intermediates in the biosynthesis of processed nuclear mRNA and cytoplasmic apoB mRNA. The proportion of apoB mRNA molecules that will be edited, have been edited by the time nuclear mRNA is polyadenylated and spliced.; http://www.haroldsmithlab.com/
![]()
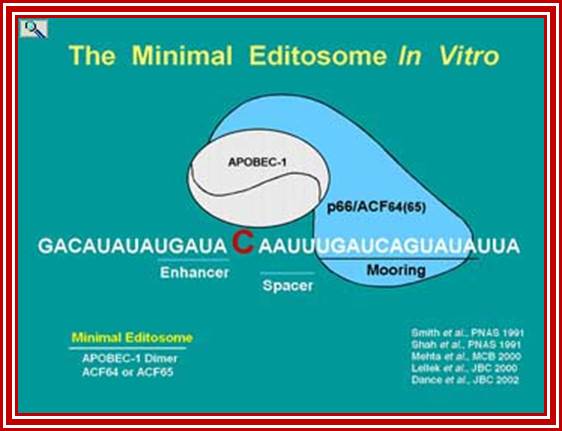
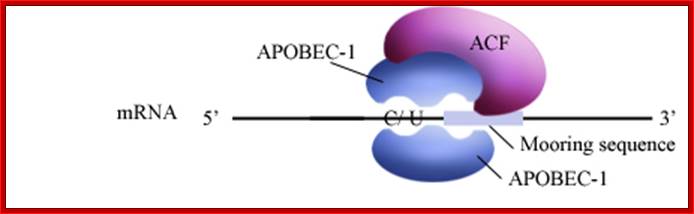
APOBEC-1 editing of apolipoprotein B mRNA; Editosome, edits C to U; http://www.haroldsmithlab.com/
ApoB mRNA editing is a posttranscriptional and post-splicing event. After splicing and polyadenylation the mRNA is edited by a simple hydrolytic cytidine deamination to uridine; and it is carried out by the APOBEC-1 enzyme, along with a complementing factor, ACF. The editing of Apo-B mRNA involves the site-specific deamination of (C 666 to U), which converts codon 2153 from a glutamine codon CAA, to a premature stop codon, UAA. ACFs are distributed in a variety of tissues, and these genes contain multiple family members. The editing complex can be represented as APOBEC-1-ACF complex.
ApoB protein transports lipid in the blood stream. Lipase in the serum will digest the triglycerides of VLDL converting them to cholesterol and protein rich LDL, a risk factor for atherosclerosis. This takes time and only occurs on VLDL assembled on ApoB100. ApoB48 containing VLDL (arising from edited ApoB mRNA) is cleared from the blood too rapidly to be converted to LDL and hence have the potential of transporting lipid without as much risk of contributing to disease.
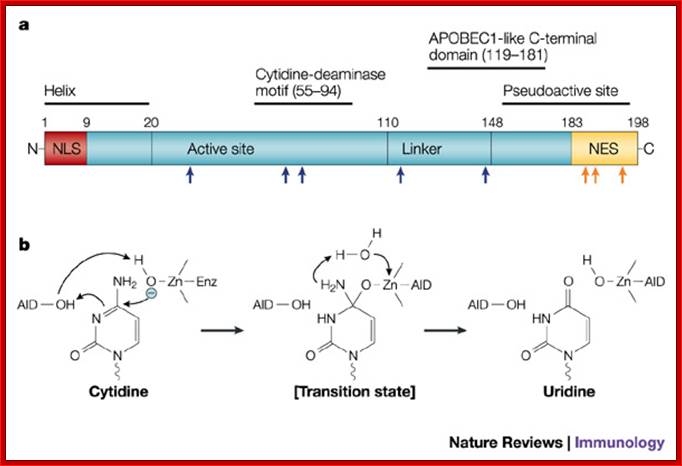
Fig. a. The primary structure of activation-induced cytidine deaminase (AID) is shown, depicting the nuclear-localization sequence (NLS), nuclear-export sequence (NES) and other predicted domains based on the structure of apolipoprotein B mRNA-editing enzyme, catalytic polypeptide 1 (APOBEC1). Some of the mutations that affect AID function are shown: mutations in the active site and linker region (blue arrows) impair both class-switch recombination (CSR) and somatic hyper mutation (SHM), whereas those at the carboxyl (C)-terminus (orange arrows) and a ten amino-acid C-terminal deletion (not shown) impair CSR without affecting SHM. N, amino terminus.
b. Mechanism of cytidine deamination,
based on a bacterial cytidine deaminase that is homologous to APOBEC1 and AID.
The reaction proceeds through a direct nucleophilic attack at position 4 of the
pyrimidine ring by zinc ions (Zn2+) coordinated with AID; Jayant Chaudhuri and Frderick W.Alt; www.Nature.com
The change of C-to-U RNA editing, in an eleven nucleotides long sequence (the mooring sequence') and the flanking nucleotides within the apolipoprotein B (apoB) mRNA are recognized by APOBEC-1 and ACF1 (APOBEC-1) complementing factor. When APOB protein complex binds on APOB mRNA, it edits the cytidine to Uridine.
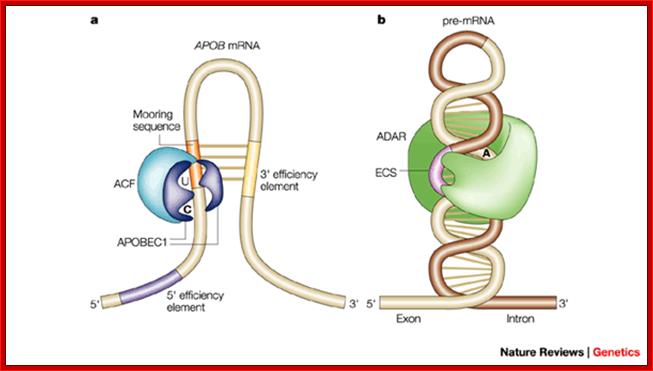
Recognition of the edited nucleotide by the editing
enzymes.a | APOBEC1 binds
to APOB mRNA in the presence of ACF and catalyses the C 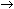 U deamination of C6666 that is positioned at the
active site. According to one model, uracil is positioned at the pseudoactive
site. It is not certain whether APOBEC1 binds as a dimer or monomer when it is
complexed with ACF. The cis-acting
sequence elements are a MOORING SEQUENCE, and 5' and 3'
efficiency elements. This is one of the many models of the configuration of
the APOB-binding site. b | ADARs recognize duplex RNA that is
formed between the editing site and the ECS that is often located in a
downstream intron. The enzymes bind to the double-stranded (ds)RNA through
their dsRBDs and deaminate a specific adenosine to inosine. ACF, APOBEC1
complementation factor; ADAR, adenosine deaminases that act on RNA; APOB,
apolipoprotein B; APOBEC1, APOB mRNA-editing enzyme catalytic
polypeptide 1; C, cytosine; ECS, editing site complementary sequence.; Liam
P. Keegan, Angela Gallo & Mary A. O'Connell;
www.Nature,com
U deamination of C6666 that is positioned at the
active site. According to one model, uracil is positioned at the pseudoactive
site. It is not certain whether APOBEC1 binds as a dimer or monomer when it is
complexed with ACF. The cis-acting
sequence elements are a MOORING SEQUENCE, and 5' and 3'
efficiency elements. This is one of the many models of the configuration of
the APOB-binding site. b | ADARs recognize duplex RNA that is
formed between the editing site and the ECS that is often located in a
downstream intron. The enzymes bind to the double-stranded (ds)RNA through
their dsRBDs and deaminate a specific adenosine to inosine. ACF, APOBEC1
complementation factor; ADAR, adenosine deaminases that act on RNA; APOB,
apolipoprotein B; APOBEC1, APOB mRNA-editing enzyme catalytic
polypeptide 1; C, cytosine; ECS, editing site complementary sequence.; Liam
P. Keegan, Angela Gallo & Mary A. O'Connell;
www.Nature,com
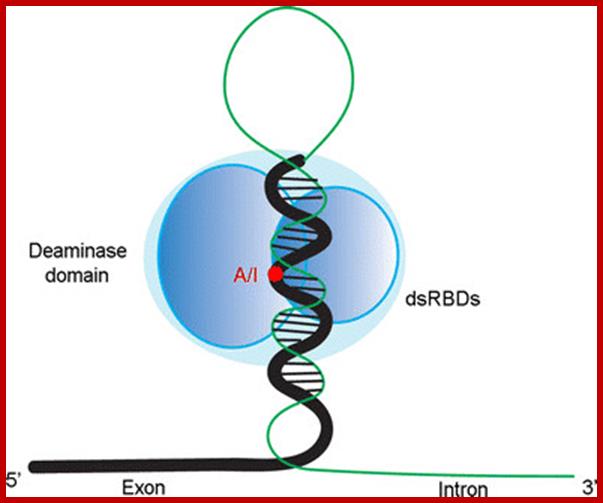
Figure: ADARs recognize duplex RNA that is formed between the editing site and the ECS site that is often located in a downstream intron.
Editing of Glutamate Receptor mRNA:
Substitution RNA editing:
In mammalian, two substitution editing sites have been found such as A-to-I and C-to-U.
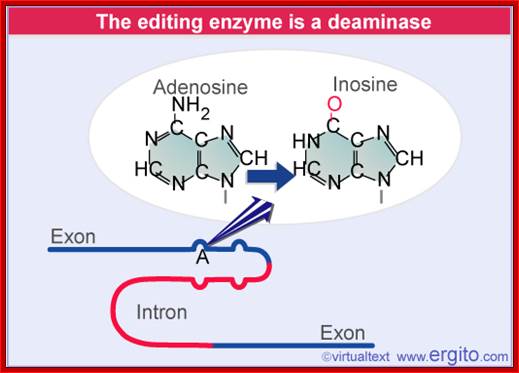
About A-to-I RNA editing, the pre-mRNA has a double-stranded RNA (dsRNA) region and the enzyme ADAR (adenosine deaminases acting on RNA) recognizes this double-stranded region. The ADAR binds to a region that is called editing site-complementary sequence (ECS) as shown in the figure. And this ADAR binding region usually contains Alu repeat patterns. When ADAR binds on ECS region, it edits a specific adenosine into Inosine.
Adenine to Inosine; http://www.nobelprize.org/
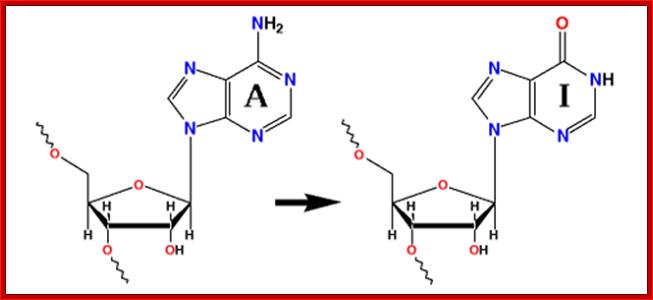
A-to-I RNA editing is catalyzed by the ADAR (adenosine deaminases) a family of enzymes and orthologues of mammalian ADARs have also been identified in D. melanogaster, C. elegans and fish species. These proteins recognize a double-stranded RNA structure formed from exonic and intronic sequences.
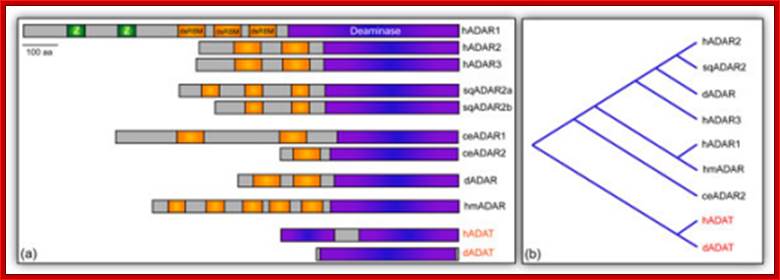
Adenosine to inosine (A-to-I) RNA editing is a post-transcriptional process by which adenosines are selectively converted to inosines in double-stranded RNA (dsRNA) substrates; ADAR family proteins; Domains of ADAR proteins; Savva YA, Rieder LE, Reenan RA - Genome Biol. (2012) http://openi.nlm.nih.gov/
The modification of RNAs by ADARs could potentially affect biological process that involves sequence or structure-specific interactions with RNA. Recent studies indicate that the editing activity of ADARs might also be involved in the antiviral interferon response pathway, where extensive deamination of viral ds-RNAs, termed hyper mutation could inhibit viral transcription and replication. Interestingly, Hepatitis Delta Virus (HDV) exploits the cellular A-to-I editing machinery to convert of a translational stop signal into a tryptophan codon within its only open reading frame. This editing event is essential for the viral life cycle since the unedited and edited protein variants have specific functions in replication and virus assembly.
Glutamate Receptor:
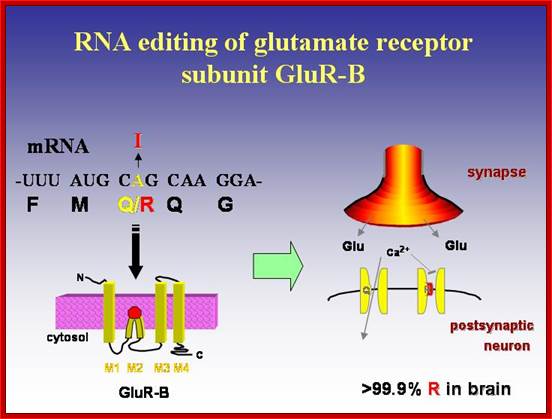
Glutamine receptor subunit GluR-B editing; http://www.uniroma2.it/
The examples of glutamate receptor subunit GluR-B RNA-editing by ADARs given above were identified as discrepancies between cDNA sequences, which are complementary to cellular mRNAs, and the corresponding genomic data. Such anecdotal evidence does not allow anyone to estimate how wide spread RNA editing is in the human genome. However, four reports were published in 2004 that evaluated A-to-I editing in the human genome and unanimously concluded that editing is very abundant indeed. Approximately 1 in every 2000 bases is edited, resulting in an estimation of more than a million edited sites in the human genome. One study identified 1637 edited genes, while a second study reported 1445 new human mRNAs that are subject to multiple editing events. The large majority of pre-mRNAs (>85%) are edited, mostly in intronic regions. One potential function for this widespread editing of introns in pre-mRNAs is to counteract the deleterious effects of double-stranded RNA, including activation of the PKR kinase and the interferon response. In addition, RNA-editing of introns may play a critical role in evolution (see Herbert, 1996).
Glutamate receptor-B is an ion channel protein which is found in all tissues. They transport both calcium and sodium ions in response to glutamate ions. In brain tissues, in specific neuronal cells, the codon CAG coding for glutamine (Gln) is changed into CIG, which codes for Arginine. Here adenine is changed to inosine by deamination and Inosine can base pair with cytosine; this leads to change in amino acid sequence and function. This change is brought about by specific enzyme called adenosine deaminase. This specific act results in the change of the character of ion channel; and now it inhibits transport of calcium ions, but transports only sodium ions. This change has tremendous implications in neuronal tissues that are involved in learning process. Absence of this kind of editing causes serious damage to the brain cells.
Genes IV; The complex may recognize a particular region of secondary structure in a manner analogous to tRNA-modifying enzymes or could directly recognize a nucleotide sequence. In the case of the GluR-B RNA, a base-paired region that is necessary for recognition of the target site is formed between the edited region in the exon and a complementary sequence in the downstream intron. A pattern of mispairing within the duplex region is necessary for specific recognition. So different editing systems may have different types of requirements for sequence specificity in their substrates; A to I conversion requires double stranded region of the RNA; http://genes.atspace.org/;
Change in the information coded by DNA occurs in mammalian and avian B-lymphocytes in generating new sequences coding for Immunoglobulins, by somatic recombination.
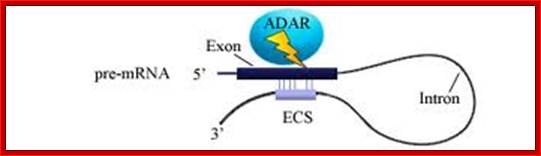
In this figure, pre-mRNA containing Alu repeat form dsRNA structure. And ADAR binding on dsRNA region edits some of the adenosines into inosines. http://openwetware.org/
The glutamate receptor mRNA where single nucleotide change, changes single amino acid which alters gating behavior and kinetic properties of the ion channels and a serotonin receptor subunit where editing regulates G-protein coupling efficiency.
The power of RNA editing in the generation of protein diversity lies in the fact that usually both the edited and unedited versions of the protein are co-expressed in the same cell and the ratio between the unedited and multiple edited variants can be regulated in a cell type-specific or time-dependent manner. Furthermore, editing within non-coding regions of pre-mRNAs can lead to alternative splicing or might affect the translation, transport or stability of RNAs. Figure 1 depicts where in primary gene transcripts A-to-I editing has been observed to date with the known or proposed sequels for the modified RNA.
Transgenic mice with even slightly reduced GluR-B editing suffer severe epileptic seizures and die within 2 weeks of age. This phenotype was a consequence of the increased Ca2+-permeability of the under edited glutamate receptors. The same phenotype resulted when the editing enzyme ADAR2 was inactivated in mice, due to a dramatic reduction in Q/R-site editing.
Other neuronal genes affected by A-to-I RNA editing include the glutamate receptor subunits GluR-C, -D, -5, and -6 where RNA editing regulates gating and kinetic properties of the ion channels, and the 5HT2C serotonin receptor where editing is known to regulate the receptor’s G-protein coupling efficiency. The cell-type specific and developmental regulation of editing-levels at multiple nucleotides in these mRNAs leads to the production of an array of receptor subtypes that differ in one or several amino acids from the unedited counterparts
In addition to these cases where the editing occurs in coding regions of gene transcripts, A-to-I editing events have also been detected in noncoding regions of cellular genes and within viral RNAs after infection. In one case, the editing enzyme ADAR2 edits its own pre-mRNA creating a new splice site that leads to alternative splicing of the pre-mRNA. The editing events seen in 5’- and 3’- untranslated regions of gene transcripts, as well as the ones affecting intronic sequences, might point toward yet unexplored functional roles of editing in the regulation of transport, stability, and further processing of RNAs.
Functional changes by A→ I, RNA editing of coding sequences. Editor meets silencer: crosstalk between RNA editing and RNA interference: Azuko Nishikura
Nature Reviews/Molecular Cell Biology
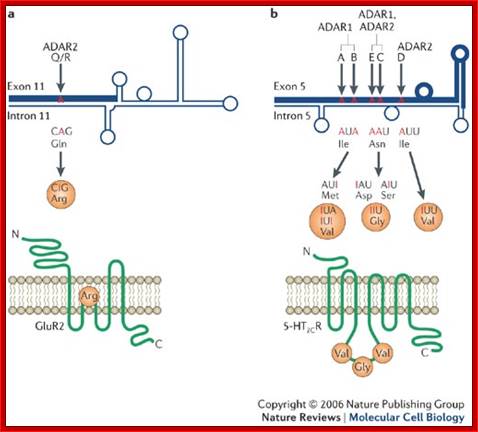
a | L-glutamate is the predominant
excitatory neurotransmitter in vertebrate nervous systems, and the glutamate
receptor (GluR) has been implicated in neuronal plasticity and higher functions
such as memory and learning. Adenosine to Inosine (A![]() I) RNA editing of the Gln/Arg (Q/R) site leads to the
replacement of a Gln by an Arg residue. Ion-channel receptors that contain the
edited GluR2 subunit are impermeable to Ca2+, whereas channels that
lack the edited subunit permit influx of Ca2+. Q/R-site editing also
regulates the tetramerization and intracellular trafficking of the receptor
protein. b | Serotonin receptors have important roles in
physiological and behavioral processes such as circadian rhythms, emotional
control and feeding behavior. G-protein-coupling functions of serotonin (5-HT)
receptor-2C (5-HT2CR) are dramatically reduced by A
I) RNA editing of the Gln/Arg (Q/R) site leads to the
replacement of a Gln by an Arg residue. Ion-channel receptors that contain the
edited GluR2 subunit are impermeable to Ca2+, whereas channels that
lack the edited subunit permit influx of Ca2+. Q/R-site editing also
regulates the tetramerization and intracellular trafficking of the receptor
protein. b | Serotonin receptors have important roles in
physiological and behavioral processes such as circadian rhythms, emotional
control and feeding behavior. G-protein-coupling functions of serotonin (5-HT)
receptor-2C (5-HT2CR) are dramatically reduced by A![]() I RNA editing that occurs at five sites (A, B, C, D and E
sites). For example, the potency of the agonist-stimulated G-protein-coupling
activity of the fully edited receptor isoform (Val-Gly-Val) is reduced by
20-fold compared with the unedited receptor isoform (Ile-Asn-Ile). The
fold-back double-stranded (ds)RNA structure, which consists of short dsRNA
regions, bulges and loops, is formed because of partial complementarity of the
exon and intronic editing-site complementary sequence (ECS; which is essential
for editing). The thick dark-blue line represents the exon, and the thin
dark-blue line represents the intron. Certain sites are exclusively edited only
by ADAR1 (adenosine deaminase acting on RNA-1) or ADAR2; ADAR2 edits
exclusively the Q/R site of GluR2 subunit and the D site of 5-HT2CR,
whereas ADAR1 selectively edits the A and B sites of 5-HT2CR. The
molecular mechanism that underlies the editing-site selectivity is not yet
completely understood. However, the secondary structure in the fold-back dsRNA
substrates, as well as functional interactions between two monomers of ADAR1 or
ADAR2, might dictate editing-site selectivity. Several intronic editing sites
that have been detected in GluR2 and 5-HT2CR dsRNAs are not shown.
I RNA editing that occurs at five sites (A, B, C, D and E
sites). For example, the potency of the agonist-stimulated G-protein-coupling
activity of the fully edited receptor isoform (Val-Gly-Val) is reduced by
20-fold compared with the unedited receptor isoform (Ile-Asn-Ile). The
fold-back double-stranded (ds)RNA structure, which consists of short dsRNA
regions, bulges and loops, is formed because of partial complementarity of the
exon and intronic editing-site complementary sequence (ECS; which is essential
for editing). The thick dark-blue line represents the exon, and the thin
dark-blue line represents the intron. Certain sites are exclusively edited only
by ADAR1 (adenosine deaminase acting on RNA-1) or ADAR2; ADAR2 edits
exclusively the Q/R site of GluR2 subunit and the D site of 5-HT2CR,
whereas ADAR1 selectively edits the A and B sites of 5-HT2CR. The
molecular mechanism that underlies the editing-site selectivity is not yet
completely understood. However, the secondary structure in the fold-back dsRNA
substrates, as well as functional interactions between two monomers of ADAR1 or
ADAR2, might dictate editing-site selectivity. Several intronic editing sites
that have been detected in GluR2 and 5-HT2CR dsRNAs are not shown.
Basically Glutamate receptors are synaptic receptors located in Neuronal membranes, very abundant in human brains. Glutamate receptors are responsible for the glutamate-mediated postsynaptic excitation of neural cells, and are important for neural communication, memory formation, learning, and regulation. Defect in editing of Glutamate receptor mRNA can lead to variety of neuronal disease including Epilepsy.
Editing by Editosomes:
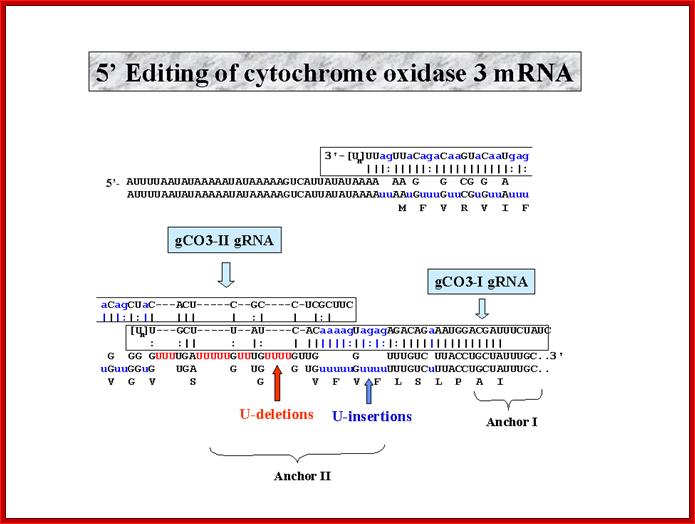
· In Trypanosomes and Leishmania, the mitochondrial transcripts, for Cytochrome oxidase II and Cytochrome oxidase III, have many missing nucleotides, especially of Uridine.
· The Cytochrome oxidase gene for subunit II consists of a frame shift of (-) 1 nucleotide, but the transcript has an additional four Us in the vicinity of the frame shift, thus it has one additional codon equivalent to one additional amino acid.
· Similar changes have been observed in SV5 measles Paramyxo viruses, involving addition of G residues into their mRNAs, which is not found in its genomic sequence.
· In Cyt-oxidase subunit-II of T. brucei, more than half of its mRNA sequences are uridines, which sequences are not found in its genomic DNA.
· In Leishmania, a parasite produces specific, but important transcripts, which are not functional, because of defects either in terms of missing nucleotides or the presence of additional nucleotides.
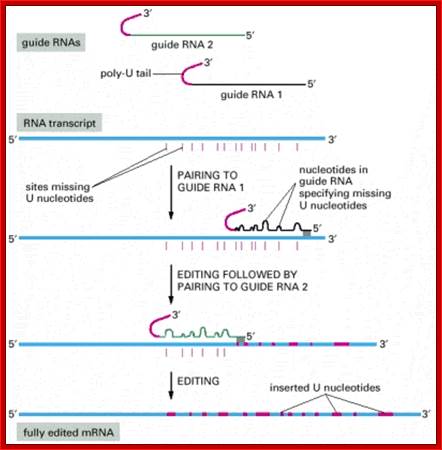
Guide RNAs:
· The parasite also contain genes coding for sequences, which are used for correcting defective pre-edited mRNAs, such transcripts are called internal guide RNAs.
· The Cyt.B oxidase transcript is added with 8 Uridine ntds wherever U nucleotides found missing at specific sites.
· The guide RNA, containing correct nucleotides as complementary sequences, acts as a catalyst and restore transcripts to be functional molecules.
· To properly edit and provide correct mRNAs editing may requires more than one IGRs (Internal Guide-RNAs).
Discovery of Guide RNAs:
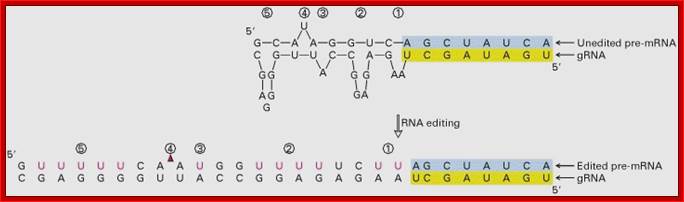
The origin of the sequence information for this insertion and deletion of U's into coding regions of mRNAs was a real mystery, as was the genetic role of the kDNA minicircles. In 1989 a solution to this puzzle was proposed. Blum et al. discovered that a novel class of small RNA molecules, transcribed both from the maxi circle and the thousands of minicircles, contained the editing information. The gRNAs were discovered by a computer search for short maxi circle sequences complementary to edited mRNA sequences. As shown below, the best hits had multiple mismatches, as shown below for the ND7 edited sequence.
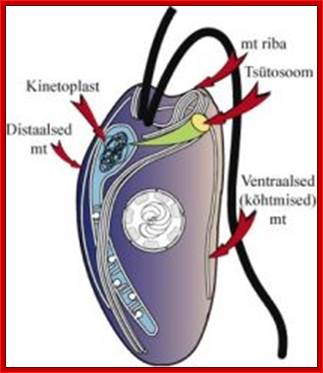
Kinetoplasts are found only in Kinetoplast flagellates.
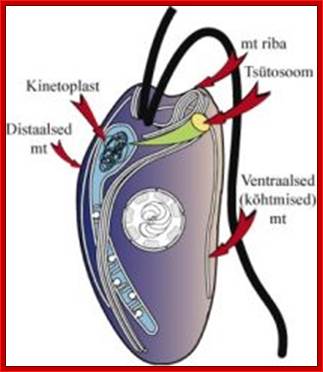
1)
long tubular mitochondria containing
kinetoplast,
2) kinetoplast - disc shaped DNA in minicircles and maxicircles (mitochondrial
DNA) located beside but not connect to kinetosome at base of flagella, 3)
kinetosome - basal body of flagella made of axoneme, 4) cytoskeleton, 5)
paraxial r,od 6) undulating membrane, 7) glycocalyx, 8) glycosomes- By secros 2010; http://www.cram.com/
When the DNA of Trypanosome mitochondrion is extracted, it does not form a simple circle typical of most mitochondria, but is a tangled collection of small loops all linked to a loop of DNA called KDNA. The structure is made up of a network of concatenated circular DNA molecules and their associated structural proteins along with DNA and RNA polymerases. The kinetoplast is found at the base of a cell's flagella and is associated to the flagellum basal body by a cytoskeletal structure; wikipedia; anisostarcentral.mbl.edu.; http://starcentral.mbl.edu/eutree_workshop/protistiary/biologyof/euglenozoa/eugzoahtmls/eugzoa_kinetop.htm

Note that all of the mismatches are "transitions"
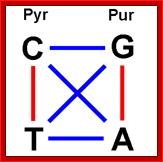
Transitions are in red and transversions in blue. http://dna.kdna.ucla.edu/.
It was realized that if the short maxicircle sequences were genes that were transcribed into RNA and if we allowed G-U base pairs (which are found in rRNAs and tRNAs), then the RNAs would form perfect duplexes with the edited sequences! We called these putative RNAs guide RNAs since they would guide the editing of the mRNA.

To prove the existence of these putative RNAs, DNA oligos were synthesized complementary to the predicted sequences and used for primer extension analysis and also by Northern analysis. By primer extension, the gRNAs had a single 5' extension product indicating a unique 5' end for each of the RNAs. Several specific gRNAs were hybrid-selected and 5' end labeled with GTP (the 5' end has a tri or di-nucleotide that can be capped with GTP using guanylyl-transferase). As shown below, there were multiple bands, indicating a 3' end heterogeneity. This was due to the presence of multiple U residues presumably added to the 3' end of the gRNA after transcription by the previously identified mitochondrial 3' terminal uridylyl transferase.
The genomic location of all Maxicircle-encoded gRNAs from Leishmania tarantula
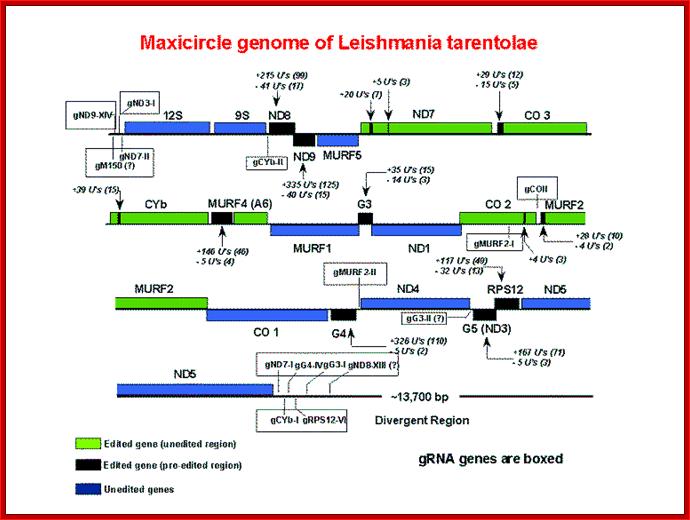
Maxicircle genome of Leishmania tarantula containing guide RNA genes; http://dna.kdna.ucla.edu/
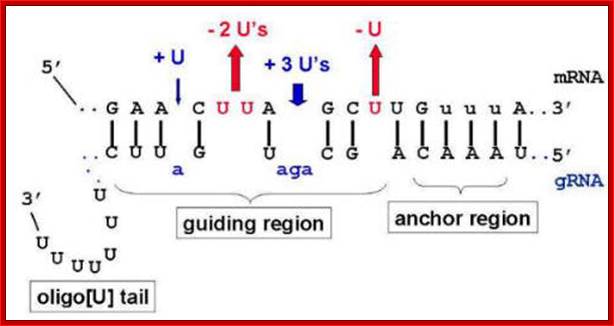
Editing of RNA in in Trypanosoma cruzi; http://dna.kdna.ucla.edu/
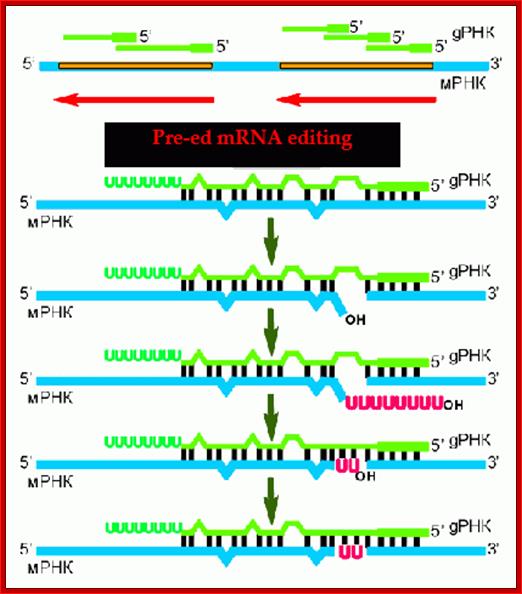
This is the summary of events that lead to editing of a pre-ed mRNA blue color and the guide RNA in green color. blogs.newsurrytheatre.org/
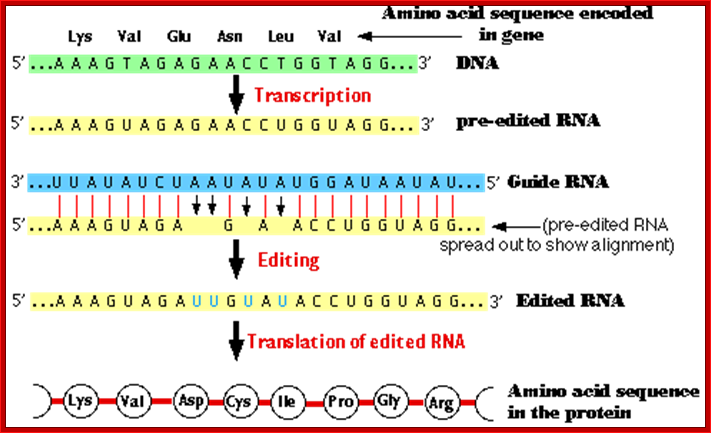
RNA editing; http://users.rcn.com; jkimball.ma. ultranet/http://www.biology-pages.info/ RNA_Editing.html
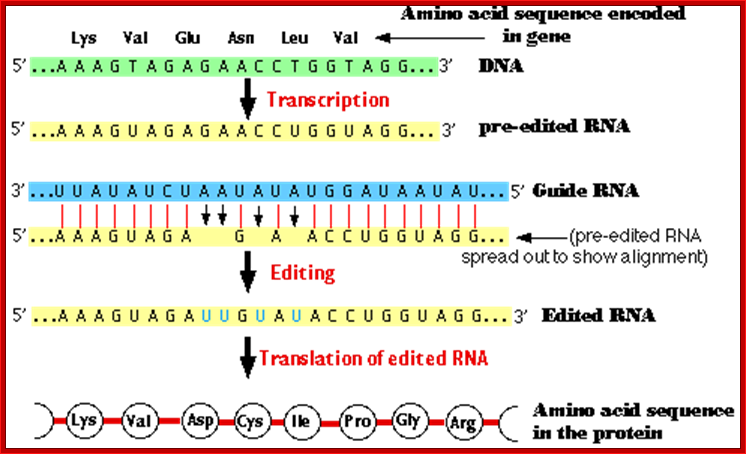
· Pre edited mitochondrial mRNA of Cyt. Oxidase subunit-II, from Trypanosome, has the following sequence; actually there is frame shift by -1 ntd.
5’---AAA GUA GA . . G. A . A C CUG GUA GG -Pre edited mRNA
· This transcript cannot produce a functional protein.
· But this frame shift change is corrected by another RNA coded for by DNA in another region, and it is called, IG-RNA Internal guide RNA.
· The IG-RNA contains a stretch of nucleotides, which are absolutely correct sequences but found as complementary sequences.
· The IG-RNA partially base pairs with pre-edited mRNAs. The 5’ end of a specific guide RNA, first base pairs with the pre edited mRNA from its 3’ end up to a region where frame shift has occurred, and then it adds an additional four Uridine residues in the vicinity of frame shift.
3’ AAA GUA GA . . G. A . A C CUGGUA GG 5’-Pre edited mRNA
5’---AAA GUA GAU UG U AUA CCU GGU –-3’Edited mRNA
· Missing U in the pre-edited mRNA is shown in red dot. Four added Us are shown in red. This mRNA is functional.
· In T. brucei COX III has more than half of the mRNA contains uridines. The edited mRNA for Cox III contains not only additional Us but also some deletions.
· Leishmania Cyt.B gene has one pre-edited gene and two guide RNA segments coded in different orientation.
Cyt b-2 (IG.RNA) Cyt-B pre edit Cyt B-1 (G.RNA)
I<-----------------I----------I------------------>I----------------------I<------
Pre-edited mRNA: 5’----AAAAGA AA . . . A. G. G. C. UUUAACC-----
Guide RNA : 3’----UUUUCUUUAAA A AA UAC AAAAUUGG.’-
Edited mRNA----AAAGAAAUUUAUGUGUCUUUUAAACCC-----3’
· The dots in pre-edit mRNA are missing Us.
· In the guide RNA the green colored are actually the nucleotides to be added to their proper positions in pre-edited RNA.
· The guide RNAs are of short length.
· The IG-RNAs 5’ end sequences match perfectly to the 3’end of the pre-edited mRNA, and when it base pairs, the IG-RNA unmatched ntds loop out.
· A specific group of enzymes which are associated with the guide RNAs help in pairing and adding nucleotides into the sites of missing nucleotides, however they add nucleotides into pre-ed mRNAs which are complementary to the nucleotides in IGR, that is if the nucleotide in the guide RNA is, A the nucleotide added into the pre-ed RNA is complementary to it i.e. U
· This way they can add nucleotides and also remove nucleotides.
· In some case more than 40 to 50% of the total numbers of nucleotides are changed in a single transcript.
· Such corrections are possible because the genome is provided with segments of DNA with correct sequences for the defective gene segment. Such gene segments are transcribed as IG-RNAs. For each of the pre-edited transcript there can be two or more IG-RNAs.
· The cell is also provided with the required protein components.
· The guide RNA and protein complexes, which perform editing, are called Editosomes. The composition of Editosomes, a 20s complex, are an endonuclease, an RNA ligase and one Uridinyl transferase.
· If the pre edited RNA requires a longer number of nucleotides to be added, more than one guide RNA is employed.
· In all cases the editing of pre-ed RNA starts from its 3’end and progressively moves towards its 5’end. For this the guide RNA associates with mRNA using anti parallel base pairing mechanism.
· The mechanism looks little elaborate to a perfectionist process.
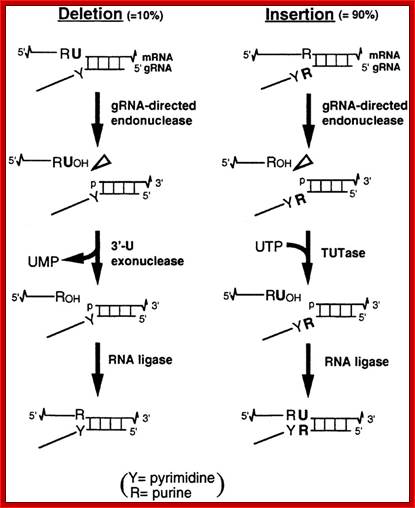
When the guide RNA is base paired to pre-ed mRNA, the unpaired ntds loop out against the missing nucleotides in the mRNA.
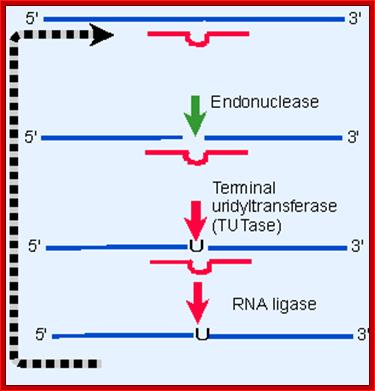
· Exactly opposite to the looped out site, the endonuclease cuts the phosphodiester bond in the pre-ed mRNA to generate a gap with 3’OH at one side and at the other side a 5’phosphate.
· The terminal uridylyl transferase (TuTase) transfers Uridine against the Adenine nucleotide in the guide RNA. Once the U is base paired to A found in IGR, the ligase joins them by covalent bond formation. In this a nucleotides is added. It is the nucleotide sequences found in the IGR are used for correcting the mistakes.
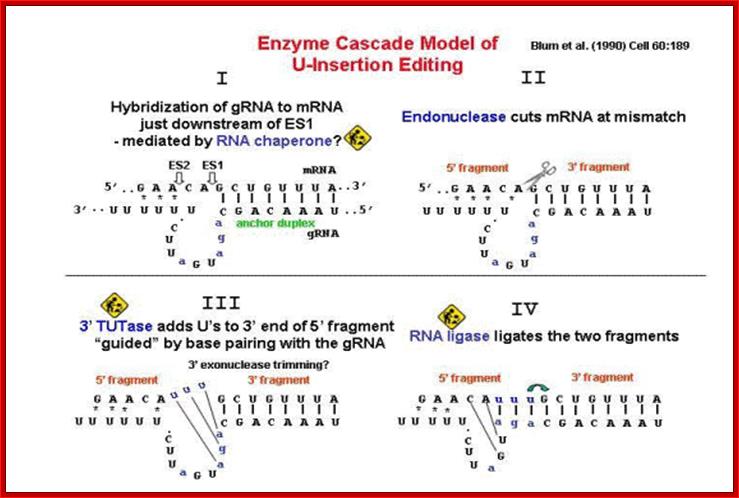
The Biochemistry of RNA editing, An additional activity, a U-specific 3'-5' exonuclease, was predicted to to be involved in the deletion of U's in U-deletion editing. A 3' TUTase and an RNA ligase activity were actually discovered to exist in trypanosome mitochondria prior to the development of this model. The ligase activity was found to be due to two proteins of approximately 45 and 50 kDa, which could be adenylated with α-32P-ATP and deadenylated by adding a ligatable RNA substrate. These proteins were components of a high molecular weight complex that sedimented around 20S in glycerol gradients. In vitro U-insertion and U-deletional activities also sedimented at around 20S, suggesting that editing was occurring in a high MW complex or complexes.; IN 2001, three labs reported the identification of the two RNA ligase genes from T. brucei.;http://dna.kdna.ucla.edu/
The structural landscape of native Editosomes in African Trypanosomes
Advanced Review: H. Ulrich Göringer, Venkata Subbaraju KATARI, CORDULA Böhm
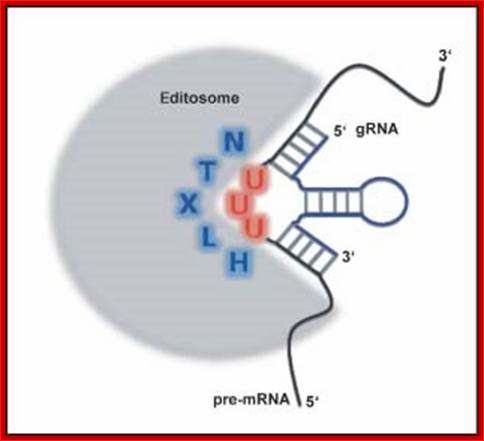
Editosomes in African Trypanosomes; http://onlinelibrary.wiley.com/

Trypanosome- Editosome of 40s size; Prof. Dr. H. Ulrich Göringer; Molecular Genetics; http://www.bio.tu-darmstadt.de/
The majority of mitochondrial pre‐messenger RNAs in African trypanosomes are substrates of a U‐nucleotide‐specific insertion/deletion‐type RNA editing reaction. The process converts nonfunctional pre‐mRNAs into translation‐competent molecules and can generate protein diversity by alternative editing. High molecular mass protein complexes termed editosomes catalyze the processing reaction. They stably interact with pre‐edited mRNAs and small noncoding RNAs, known as guide RNAs (gRNAs), which act as templates in the reaction. Editosomes provide a molecular surface for the individual steps of the catalytic reaction cycle and although the protein inventory of the complexes has been studied in detail, a structural analysis of the processing machinery has only recently been accomplished. Electron microscopy in combination with single particle reconstruction techniques has shown that steady state isolates of editosomes contain ensembles of two classes of stable complexes with calculated apparent hydrodynamic sizes of 20S and 35–40S. 20S editosomes are free of substrate RNAs, whereas 35–40S editosomes are associated with endogenous mRNA and gRNA molecules. Both complexes are characterized by a diverse structural landscape, which include complexes that lack or possess defined subdomains. Here, we summarize the consensus models and structural landmarks of both complexes. We correlate structural features with functional characteristics and provide an outlook into dynamic aspects of the editing reaction cycle. WIREs RNA 2010 DOI: 10.1002/wrna.67.
RNA Editing Hints of a Remarkable Diversity in Gene Expression Pathways, Scott D. Siewert.
“In The Rescue: A Romance in the Shallows, Joseph Conrad stoically proclaims "there is no rest for the messenger till the message is delivered." Although he could not have known, Conrad's statement serves as an apt metaphor for the dramatic remodeling of precursor mRNAs (pre-mRNAs) by the now familiar RNA processing reactions of higher eukaryotes. Although less explored, reactions required for gene expression in early diverging eukaryotes have demonstrated the generality of this metaphor within the eukaryotic kingdom.
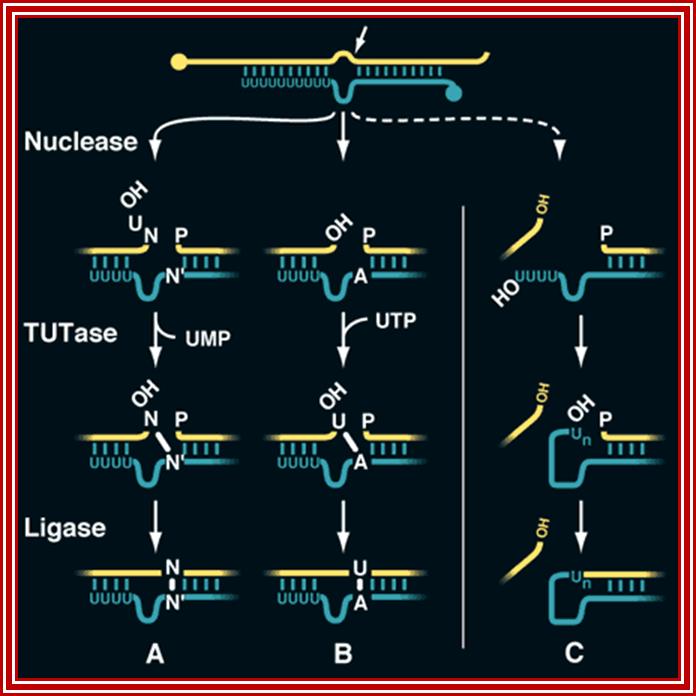
A mechanism for RNA editing. Uridylate (A) deletion, (B) insertion, and (C) chimera formation occur through similar mechanisms. Chimeras are formed when an interaction between the 3'-oligo(U) tail of the gRNA and the 5'-cleavage product is weakened. For clarity, the portions of the pre-mRNA (yellow) and the gRNA (green) not adjacent to the processing site are omitted.; Scott D. Siewert; http://www.sciencemag.org/
A mechanism for RNA editing: Uridylate (A) deletion, (B) insertion, and (C) chimera formation occur through similar mechanisms. Chimeras are formed when an interaction between the 3'-oligo (U) tail of the gRNA and the 5' -cleavage product is weakened. For clarity, the portions of the pre-mRNA (yellow) and the gRNA (green) not adjacent to the processing site are omitted.
Mitochondrial DNA in trypanosomes is unusual in that it comprises two classes of circular molecules. The primary transcripts derived from the larger class of DNAs, maxicircles, have their coding information remodeled by the site-specific insertion and deletion of uridylate (U) residues. This sequence remodeling creates mRNAs homologous to those encoded by the mitochondrial DNA in other organisms and often produces more than 50% of a transcript's coding information
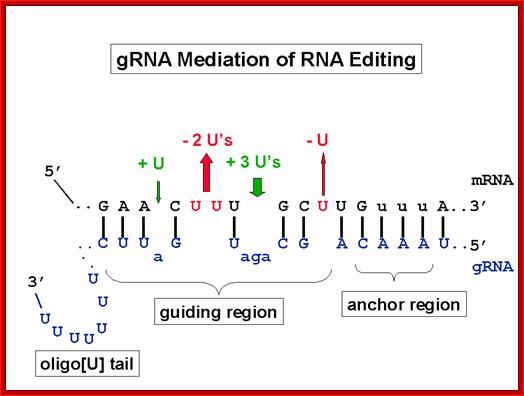
These small RNAs, termed guide RNAs (gRNAs), have a sequence of approximately 10 nucleotides at their 5' end that allows them to hybridize to a specific pre-mRNA at a defined location. Bulged purine nucleotides in the adjacent sequence of the gRNA are capable of directing the appropriate insertion of U's into the pre-mRNA, and U's bulged in the pre-mRNA could be deleted. Both of these events would extend the complementarity of the initial intermolecular duplex to produce a pre-mRNA containing the mature sequence. Thus, RNA editing could involve the transfer of genetic information from one RNA to another.
One popular model suggested that RNA editing is mechanistically related to the self-splicing reactions of introns. In this model, non-encoded U residues at the 3' end of gRNAs serve as a repository for those inserted and deleted within pre-mRNAs during two coupled transesterification reactions. Excitement grew for this proposal after the discovery of chimeric molecules consisting of 5' gRNA sequences linked to the 3' portion of their cognate pre-mRNAs, the intermediates predicted by this model.
Formal proof of the reaction mechanism required biochemical probing of the RNA editing reaction, studies that were made possible in part by my thesis work. Using an assay system that indirectly monitored processing, I was able to demonstrate the transfer of genetic information from gRNA to pre-mRNA in vitro thus confirming that gRNA is the source of genetic information during the RNA editing reaction”.
![]()
Addition or deletion of U residues for that matter any residue dictated by its guide RNA; http://genes.atspace.org/
gRNA mediated editing; http://dna.kdna.ucla.edu/
Pairing of guide RNA to unedited mRNA; edrna.mbc.nctu.edu.tw
http://dna.kdna.ucla.edu/
RNA editing in the mitochondria of trypanosomes: Guide RNAs contain at their 3′ end a stretch of poly U, which donates U nucleotides to sites on the RNA transcript that mis pair with the guide RNA; thus, the poly-U tail gets shorter as editing proceeds; www.sakshieducation.com
Deletional editing
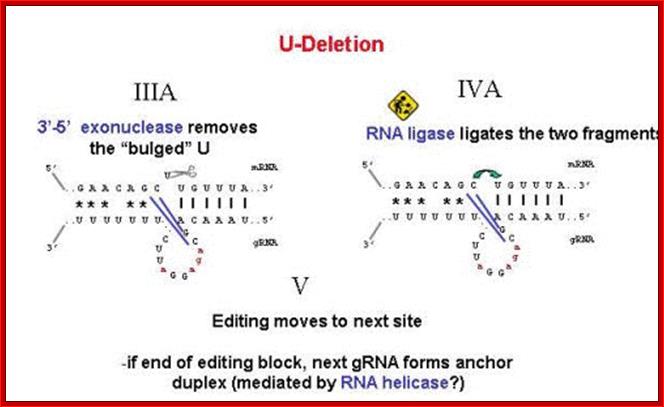
· The editing process which involves in the removal a nucleotide from the pre-ed RNA, the internal guide RNA base pairs extensively leaving the regions or nucleotide in the pre-ed mRNA looped out, where there is no complement arity in IGR.
· Any nucleotide(s) in pre-ed RNA that is looped out against a nucleotide in
the guide RNA, an endonuclease cuts at the 3’ side of the looped out nucleotide, the nucleotide is U ntd, thus it generates U with free 3’OH group and the adjacent nucleotide with 5’ phosphate group.
· An exonuclease chops out or removes the looped out nucleotide from 3’ end, in this case it is Uridine, thus it generates another 3’OH group.
· By this unique mechanism, in T. brucei; CO III mRNA is 731 ntd long, and 407 nucleotides to the functional mRNA and 19 encoded uridines are deleted.
Roles for ligases in the RNA editing complex of Trypanosoma brucei: band IV is needed for U-deletion and RNA repair,
Trypanosome RNA Editing: Simple Guide RNA Features Enhance U Deletion 100-Fold; Current understanding of U deletion and U insertion. RNA editing is catalyzed by a complex with seven major polypeptides, two of which correspond to distinct ligases (12, 29). U deletion and U insertion involve gRNA-directed endonuclease cleavage of the pre-mRNA (shown by an arrowhead), either 3′-U-exo or TUTase acting on the upstream fragment, and RNA ligase rejoining the mRNA (10, 17, 32). R, purine(s); Y, pyrimidine(s). These two forms of editing use distinct cleavage activities (11) and distinct ligase enzymes (Cruz-Reyes et al., unpublished), and the 3′-U-exo is not a reverse TUTase reaction (10, 29), which is indicated by the unequal sign. gRNAs have three main portions: a 5′ region to anchor the gRNA to the mRNA just downstream of the editing site and direct the cleavage, a central region to guide the U deletions and insertions at mismatches in base pairing with the pre-mRNA, and a 3′ oligo(U) that may tether the very purine-rich upstream pre-mRNA. Many additional conserved gRNA features have also been noted (see the text). Dotted lines indicate potential base pairs that could serve in a ligation; Catherine E. Huang, Jorge Cruz-Reyes, Alevtina G. Zhelonkina, Sean O'Hearn, Elizabeth Wirtz and Barbara Sollner-Webb
Kinetoplastid DNA
Kinetoplast DNA is relatively abundant and consists of mini-circles and maxi-circles. The two types of ktDNA occur in a concatenated mass within the mitochondria. Maxi-circles encode several mitochondrial genes and are more-or-less equivalent to the mtDNA. Mini-circles are heterogeneous and rapidly evolving and their function is less clear. Both mini-circles and maxi-circles encode guide RNA genes. Some genes on the maxi-circles have 'errors' which need to be edited. The guide RNAs are important for this RNA editing that takes place in the mitochondria of kinetoplasts. The editing of these 'crypto genes' is believed to occur in a hypothetical 'Editosome' particle. The extent of editing seems to correlate with different parasite life cycle stages and the corresponding changes in metabolism (i.e., aerobic vs. anaerobic) that are associated with the different life cycle stages. Mini-circle DNA is also used for parasite detection and distinguishing different isolates. http://www.tulane.edu/
The major distinguishing feature of this group is a subcellular structure known as the kinetoplast. The kinetoplast is a dark Giemsa-staining structure which is distinct from the nucleus (Figure). The size of the kinetoplast will vary according to species. The kinetoplast is found near the basal body which is located at the base of the flagellum (Figure). Because of this location near the flagellum, it was previously believed that the kinetoplast was somehow associated with cell movement--hence the name. However, the kinetoplast is actually a distinct region of the mitochondria and is not involved in motility. The staining of the Kinetoplastid is due to mitochondrial DNA (see Box). In fact, the existence of extranuclear (i.e., organellar) DNA was first demonstrated in the Kinetoplastids.
Kinetoplast DNA containing guide RNA for mitochondrial RNA editing

Kinetoplastid DNA- Maxi and minicircles; www.Primery RNA transcript’ sasec.in/small images
http://users.rcn.com/
· In leishmania brucei, Kinetoplast maxi circle DNAs is catenated with mini-circle DNAs. The Kinetoplast DNA is nothing but mitochondrial DNA.
· Guide RNAs for each pre-ed mRNA transcripts are encoded in not only max-circles and also in minicircles.
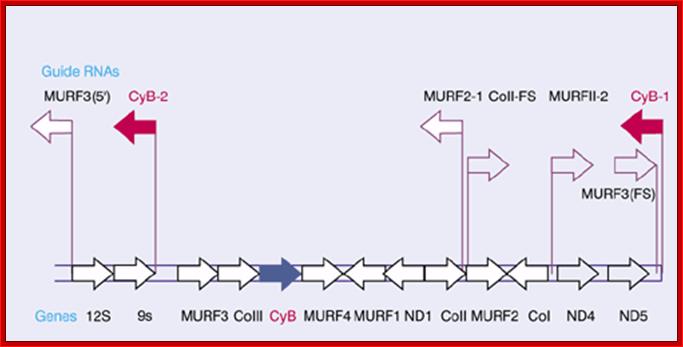
The guide RNAs (IG-RNAs) are found for the following genes-
<MuRF3’, 12S >, 9S >, < Cyt.b-2 , MuRF3>, Co III>, Cyb >, MuRF4> >,< MuRF1, < InD1, CoII >, < MURF 2-1, CoII >>, MuRF2 >, < Co I, MuRFII>, ND4>, ND5>, < Cyb I . Arrows indicate the direction of transcription. URF stands for unidentified reading frame; Guide RNA genes are clustered and their orientation of them has depicted. http://genes.atspace.org/
Impact of A-I editing;
A to I-editing occurs in regions of double stranded RNA (dsRNA). A to I editing can be specific (a single adenosine is edited within the stretch of dsRNA) or promiscuous (up to 50% of the adenosines are edited). Specific editing occurs within short duplexes (e.g. those formed in an mRNA where intronic sequence base pairs with a complementary exonic sequence), while promiscuous editing occurs within longer regions of duplex (e.g. pre- or pri-miRNAs, duplexes arising from transgene or viral expression, duplexes arising from paired repetitive elements). There are many effects of A to I editing, arising from the fact that I behaves as if it is G both in translation and when forming secondary structures. These effects include alteration of coding capacity, altered miRNA or siRNA target populations, heterochromatin formation, nuclear sequestration, cytoplasmic sequestration, Endo nucleolytic cleavage by Tudor-SN, inhibition of miRNA and siRNA processing and altered splicing.]
RNA editing in plants
It has been shown in previous studies that the only types of RNA editing seen in the plants’ mitochondria and plastids are conversion of C to U and U to C (very rare). RNA editing sites are mainly found in the coding regions of mRNA, introns, and other non-translated regions. In fact, RNA editing can restore the functionality of tRNA molecules. The editing sites are primarily found upstream of mitochondrial or plastid RNAs. The exact mechanism is unknown, but previous studies have speculated the involvement of gRNA and the editosomal complex. The reason behind that specific idea arose from the fact that there are too many editing sites that needed to be changed in those organelles for a deaminase.
RNA editing is essential for the normal functioning of the plant’s translation and respiration activity. Editing can restore the essential base-pairing sequences of tRNAs, restoring functionality. It has also been linked to the production of RNA edited proteins that are incorporated into the polypeptide complexes of the respiration pathway. Therefore, it is highly probable that polypeptides synthesized from unedited RNAs would not function properly and hinder the activity of both mitochondria and plastids.
RNA editing in animals:
The first observance of RNA editing in the animal mitochondrial is poly-A addition. Polyadenylation is responsible for the derivation of the 3’ termini of numerous mRNAs in animals. The 3’ termini are generated from the processing of the 5’ ends of downstream tRNAs followed by limited polyadenylation of the mRNAs. In fact, some of the nascent mRNA 3’ termini do not contain a stop codon to end translation and may end with a U (after the last in-frame codon). The U is extended to generate a UAA stop codon by 3’ addition via a polyA polymerase. However, if these incomplete mRNA transcripts were not edited, translation would be terminated and the ribosome would dissociate. RNA editing is essential in the completion of some transcripts and insures proper translation of proteins in the mitochondria of animals.

RNA editing; Summary of the Various Functions of RNA Editing; http://en.wikipedia.org/
Insertion/Deletion Editing
Example: the gene (in the mitochondria of Trypanosome brucei) for one of the subunits of cytochrome C oxidase
Several genes encoded in the mitochondrial DNA of this species (the cause of sleeping sickness in humans) encode transcripts that must be edited to make the mRNA molecules that will be translated into protein.
Editing requires a special class of RNA molecules called guide RNA (gRNA).
These small molecules have sequences that are complementary to the region around the site to be edited.
The guide RNA base-pairs — as best it can — with this region.
(Note that in addition to the usual purine-pyrimidine pairing of C-G and A-U, G-U base-pairing can also occur.)
Because of the lack of precise sequence complementarity, bulges occur either
- in the guide RNA where, usually, there are As not found in the transcript to be edited (as shown here) or
- in the transcript to be edited.
The bulges are eliminated by cutting the backbone of the shorter molecule and inserting complementary bases.
- In the first case (shown here) this produces insertions (here of Us)
- In the second case (not shown) this produces deletions.
Note that in the example shown here, the insertion of 4 nucleotides has created a frameshift so that the amino acids encoded downstream (after Val) in the edited RNA are entirely different from those specified by the gene itself.
Some other examples of insertion/deletion editing:
Insertion/deletion editing has also been found to occur with -
- mRNA, rRNA, and tRNA transcripts in the mitochondria of the slime mold Physarum polycephalum.
- in measles virus transcripts.
Why RNA Editing?
Good question. Some possibilities:
- Perhaps — like alternative splicing — it is a mechanism to increase the number of different proteins available without the need to increase the number of genes in the genome. (The human genome is not that much larger than those of Drosophila and C. elegans, but our proteome is much larger.)
- So it can create proteins with slightly different functions to use in specialized circumstances.
- The ability to synthesize two versions (with different functions) of apolipoprotein B from a single gene — as shown above — is an example. Note that in this case, RNA editing has accomplished the same result as alternative splicing.
- There is evidence that Drosophila (and humans) use editing to create subtle differences in the properties of proteins in different regions of the brain.
- some voltage-gated ion channels and
- some receptors of neurotransmitters
- RNA (as well as DNA) editing may help protect the genome against
- some viruses (and against which they often evolve counter-measures)
- damage by retrotransposons
- Other possibilities: The untranslated regions at either end of many messenger RNA (mRNA) molecules — the 5'-UTRs and 3'-UTRs — often contain sequences of nucleotides that permit intramolecular base-pairing resulting in stretches of double-stranded RNA (dsRNA). (View another example.) But dsRNA in the cell runs the risk of being destroyed by RNA interference (RNAi). Perhaps RNA editing in these areas protects against that risk. There is also evidence that RNA editing (converting As to Is in the 3'-UTR) of precursor mRNAs is a signal to retain them within the nucleus ready to be quickly exported if needed by the cell.
- Or perhaps it is simply the legacy of a system that was first used to correct RNA transcripts for harmful mutations in the DNA encoding them. Now with no strong evolutionary pressure to correct the DNA, RNA editing persists.
So RNA editing appears to be here to stay. In fact, defects in RNA editing are associated with some human cancers as well as with amyotrophic lateral sclerosis (ALS — "Lou Gehrig's disease").
The complexity of higher organisms is based on the number of different gene products available for structural, enzymatic and regulatory functions. Recently, whole-genome sequencing of a diverse set of species from primitive to highly developed organisms (bacteria, yeast, worm, fly, fish, mouse and human) has revealed that during evolution the number of genes in genomes has not increased at the same rate as the observed organismal complexity. For example, the fruit fly (D. melanogaster) carries fewer genes than the nematode C. elegans (~14,000 versus ~19,000) even though the fly is obviously more complex. And when comparing these numbers from invertebrates to the estimated number of genes in the human genome (ca. 30,000), it is inconceivable that the increase in gene numbers alone could be the basis for the tremendous evolutionary increase in diversity and complexity.
These findings underscore the important role the mechanisms that control gene utilization play in the creation of proteomic and phenotypic diversity. Several post-transcriptional and post-translational mechanisms have been identified that lead to the production of multiple gene products from a single gene. The alternative splicing of pre-mRNAs is a particularly frequent event, estimated to affect more than 50% of mammalian primary transcripts.
Molecular Mechanism and substrate requirements for site-selective RNA editing:
Mechanistically, the A-to-I editing reaction follows a hydrolytic deamination mechanism catalyzed by a Zn2+ coordinating enzyme, which likely is active as a dimer and does not require accessory factors for activity. The ADARs are distantly related to DNA methyl-transferases and believed to employ a similar base-flipping mechanism for base modification. Double-stranded RNA binding domains and the catalytic region of the RNA editing enzyme convey dsRNA specific adenosine deaminase activity, which on extended perfect dsRNA proceeds promiscuously. The high site-selectivity and specificity of the RNA editing reactions involving physiological substrates is achieved in large part by the overall three dimensional structure of the imperfectly base-paired pre-mRNA with bulges and loops. However, the basis for the selectivity observed for certain RNAs is poorly understood as is the interplay between catalytic and RNA-binding domains of the editing enzymes.
To date it is not possible to predict which adenosine within a given RNA will undergo editing, or if a computer-predicted RNA secondary structure will be a competent in vivo substrate for an ADAR enzyme. When analyzing the predicted RNA secondary structures of endogenous transcripts known to be subject to A-to-I editing, the modified adenosines reside in base-paired regions, mismatched or within a loop. Mini-gene substrates and compensatory mutational analysis have been used to confirm the predicted RNA folds, however, the three-dimensional structures and interactions of RNA and protein components are not known. Through in vitro studies and analysis of the modification patterns of endogenous substrates a 5’ neighbor preference (ADAR1, 2) and 3’ neighbor preference (ADAR2) have been established.
The RNA editing mechanism causes functional activities different from the unedited transcripts. As shown in, RNA editing may alter processes including mRNA translation by changing codons; RNA editing may alter pre-mRNA splicing patterns by changing splice site recognition sequences; RNA editing affect RNA degradation by modifying RNA sequences involved in nuclease recognition; RNA editing may effect viral RNA genome stability by changing template and hence product sequences during RNA replication; and RNA editing potentially may affect RNA structure dependent activities that entail binding of RNA by proteins.
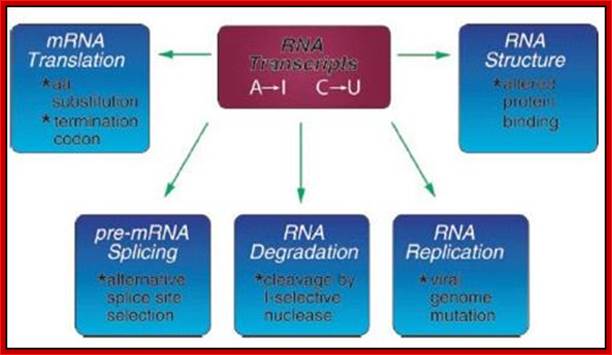
RNA Editing; http://edrna.mbc.nctu.edu.tw/
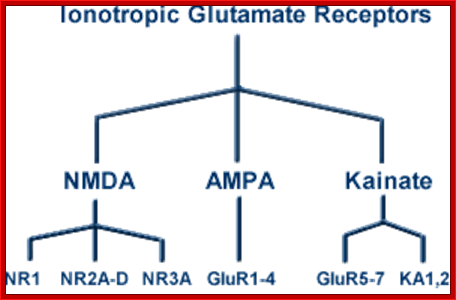
An important form of regulation appears to be achieved by editing the primary RNA transcripts of the AMPA and KA receptor subunits. For example, AMPA receptors that contain the GluR2 subunit are much less permeable to Ca2+ than those assembled without GluR2. This important feature of GluR2 was traced by site-directed mutagenesis techniques to a single amino acid within the second membrane-associated domain. A glutamine (Q) resides in this position in GluR1, GluR3 and GluR4, but an arginine (R) is present in GluR2.

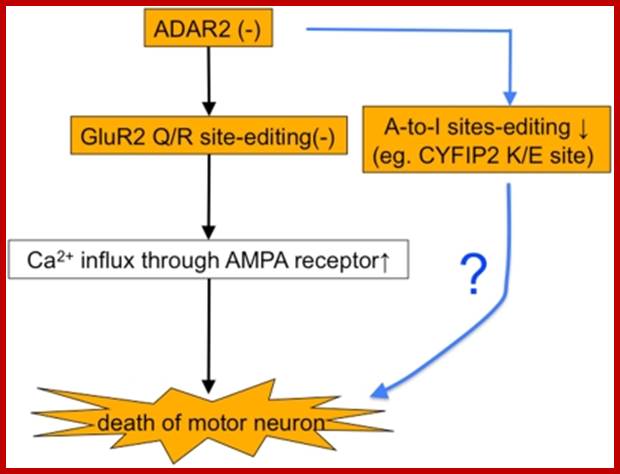
When
does ALS start? ADAR2-GluA2 hypothesis for the etiology of sporadic ALS; Amyotrophic
lateral sclerosis (ALS) is the most common adult-onset motor neuron disease.
More than 90% of ALS cases are sporadic, and the majority of sporadic ALS
patients do not carry mutations in genes causative of familial ALS; therefore,
investigation specifically targeting sporadic ALS is needed to discover the
pathogenesis. The motor neurons of sporadic ALS patients express unedited GluA2
mRNA at the Q/R site in a disease-specific and motor neuron-selective manner.
GluA2 is a subunit of the AMPA receptor, and it has a regulatory role in the
Ca(2+)-permeability of the AMPA receptor after the genomic Q codon is replaced
with the R codon in mRNA by adenosine-inosine conversion, which is mediated by
adenosine deaminase acting on RNA 2 (ADAR2). Therefore, ADAR2 activity may not
be sufficient to edit all GluA2 mRNA expressed in the motor neurons of ALS
patients. To investigate whether deficient ADAR2 activity plays pathogenic
roles in sporadic ALS, we generated genetically modified mice (AR2) in which
the ADAR2 gene was conditionally knocked out in the motor neurons. AR2 mice
showed an ALS-like phenotype with the death of ADAR2-lacking motor neurons.
Notably, the motor neurons deficient in ADAR2 survived when they expressed only
edited GluA2 in AR2/GluR-B(R/R) (AR2res) mice, in which the endogenous GluA2
alleles were replaced by the GluR-B(R) allele that encoded edited GluA2. In
heterozygous AR2 mice with only one ADAR2 allele, approximately 20% of the
spinal motor neurons expressed unedited GluA2 and underwent degeneration,
indicating that half-normal ADAR2 activity is not sufficient to edit all GluA2
expressed in motor neurons. It is likely therefore that the expression of
unedited GluA2 causes the death of motor neurons in sporadic ALS. We
hypothesize that a progressive downregulation of ADAR2 activity plays a
critical role in the pathogenesis of sporadic ALS and that the pathological
process commences when motor neurons express unedited GluA2.
When does ALS start? ADAR2-GluA2 hypothesis for the etiology of sporadic ALS
(PDF Download Available). Available from: https://www.researchgate.net/publication/51816954_When_does_ALS_start_ADAR2-GluA2_hypothesis_for_the_etiology_of_sporadic_ALS
[accessed Jun 11, 2017].
https://www.researchgate.net/publication/51816954_When_does_ALS_start_ADAR2-GluA2_hypothesis_for_the_etiology_of_sporadic_ALS [accessed Jun 11, 2017].
Thus, this site has been named the “Q/R site.” In addition to Ca2+ permeability, the Q/R site influences the single-channel conductance and the sensitivity of the activated receptor to block by polyamine spider toxins and internal polyamines. Voltage-dependent block by internal polyamines gives rise to inward rectification of AMPA receptors with low GluR2 abundance. Interestingly, the genomic DNA sequence has a glutamine codon in the Q/R position, even for subunits such as GluR2, in which the mature mRNA has an arginine codon in this site. A double-stranded RNA adenosine deaminase is employed as an “editor” to control the amino acid encoded by this critical codon [16]. RNA editing and associated changes in ionic permeability also have been demonstrated for some KA receptor subunits. The conditions under which neurons utilize RNA editing to regulate the permeability properties of their glutamate receptors remain to be demonstrated.
Given this combination of internal and C-terminal splice variants and Q/R editing, it appears that four to eight or more mature RNAs can be made from each of the 16 known genes encoding ionotropic receptor subunits. Thus, neurons have a massive degree of flexibility in constructing a potentially huge number of receptors. The actual degree of glutamate receptor heterogeneity utilized by neurons remains a major unanswered question.
The
Good and the Bad of Glutamate Receptor RNA Editing; Glutamate
receptors play a key role in excitatory synaptic transmission and plasticity in
the central nervous system (CNS). Their channel properties are largely dictated
by the subunit composition of tetrameric receptors.
Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate channels
are assembled from GluA1–4 AMPA or GluK1–5 kainate receptor subunits. However,
their functional properties are highly modulated by a post-transcriptional
mechanism called RNA editing. This process involves the enzymatic deamination
of specific adenosines (A) into inosines (I) in pre-messenger RNA. This
post-transcriptional modification leads to critical amino acid substitutions in
the receptor subunits, which induce profound alterations of the channel
properties. Three of the four AMPA and two of the five kainate receptor
subunits are subjected to RNA editing. This study reviews the advances in
understanding the importance of glutamate receptor RNA editing in finely tuning
glutamatergic neurotransmission under physiological conditions and discusses
the way in which the dis-regulation of RNA editing may be involved in neurological
pathology.
The Good and the Bad of Glutamate Receptor RNA Editing (PDF Download
Available). Available from: https://www.researchgate.net/publication/309311484_The_Good_and_the_Bad_of_Glutamate_Receptor_RNA_Editing
[accessed Jun 11, 2017]. Alice Filippini et al.
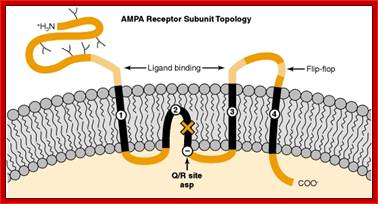
Location and role of Q/R site in ion permeation through AMPA receptors. The amino acid sequences of GluR1-GluR4 are identical within the pore loop, except for the Q/R site, as shown. The phenotypes of AMPA receptors containing edited or unedited GluR2 or lacking GluR2 are summarized. Pca, calcium permeability; γ(pS), single channel conductance
Other neuronal genes affected by A-to-I RNA editing include the glutamate receptor subunits GluR-C, -D, -5, and -6 where RNA editing regulates gating and kinetic properties of the ion channels, and the 5HT2C serotonin receptor where editing is known to regulate the receptor’s G-protein coupling efficiency. The cell-type specific and developmental regulation of editing-levels at multiple nucleotides in these mRNAs leads to the production of an array of receptor subtypes that differ in one or several amino acids from the unedited counterparts.
In addition to these cases where the editing occurs in coding regions of gene transcripts, A-to-I editing events have also been detected in noncoding regions of cellular genes and within viral RNAs after infection. In one case, the editing enzyme ADAR2 edits its own pre-mRNA creating a new splice site that leads to alternative splicing of the pre-mRNA. The editing events seen in 5’- and 3’- untranslated regions of gene transcripts, as well as the ones affecting intronic sequences, might point toward yet unexplored functional roles of editing in the regulation of transport, stability, and further processing of RNAs.]
Plants’ mitochondria too go through editing:
RNA editing in plant mitochondria is a post-transcriptional process involving the partial change of C residues into U. These C to U changes lead to the synthesis of proteins with an amino acid sequence different to that predicted from the gene. Proteins produced from edited mRNAs are more similar to those from organisms where this process is absent. This biochemical process involves cytidine deamination. The cytoplasmic male sterility (CMS) phenotype generated by the incompatibility between the nuclear and the mitochondrial genomes is an important agronomical trait which prevents inbreeding and favors hybrid production. The hypothesis that RNA editing leads to functional proteins has been proposed. This hypothesis was tested by constructing transgenic plants expressing a mitochondrial protein translated from unedited mRNA. The transgenic "unedited" protein was addressed to the mitochondria leading to the appearance of mitochondrial dysfunction and generating the male sterile phenotype in transgenic tobacco plants. Male sterile plants were also obtained by expressing specifically a bacterial ribonuclease in the anthers. The economical benefits of artificially engineered male-sterile plants or carrying the (native) spontaneous CMS phenotype, implies the restoration to obtain fertile hybrids that will be used in agriculture. Restoration to fertility of transgenic plants was obtained either by crossing male-sterile plants carrying the "unedited" mRNA with plants carrying the same RNA, but in the antisense orientation or, in the case of plants expressing the ribonuclease, by crossing male-sterile plants with plants expressing an inhibitor specific of this enzyme.


Phenotype due to editing; Transgenic plants carrying the edited and unedited forms of atp9 were shown to have integrated the gene at the nuclear level and to express atp9at the transcriptional and translational level. The atp9 protein was shown to be localized in the mitochondrial fraction of transgenic tobacco plant. The atp9 protein was shown to be localized in the mitochondrial fraction of transgenic tobacco; Michel Hemould, Amand Mouras and Simon Lifvak;http://www.scielo.cl/
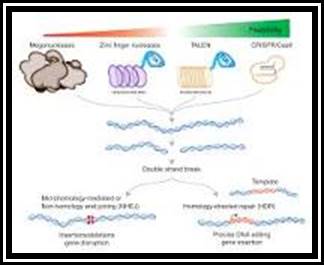
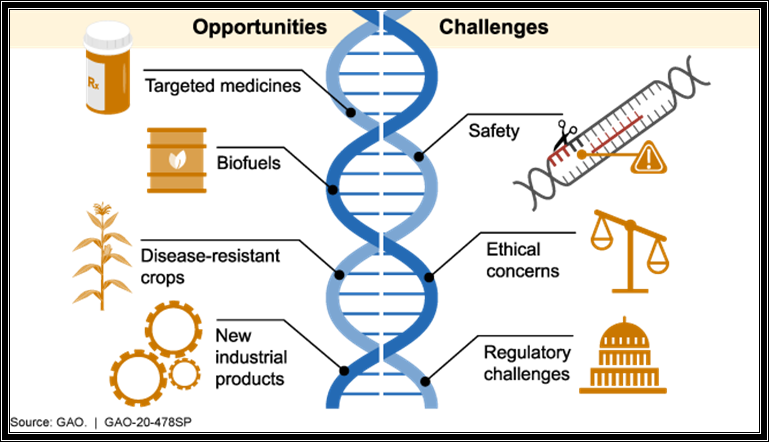
10 Years ago, it was first proposed in the journal Science that CRISPR (clustered regularly interspaced short palindromic repeats)/CRISPR associated systems (Cas) could be used for targeted gene editing by creating specific double stranded breaks in target DNA [1]. In 2020, Dr Emmanuelle Charpentier and Dr Jennifer Doudna won the Nobel prize in Chemistry for their work. Since the discovery CRISPR has allowed for advancements within research into genome editing: making processes faster, cheaper, and more accurate [2].
In prokaryotes, CRISPR is an RNA-mediated adaptive immune response used by the cell to target invading viruses, bacteriophages, and plasmids. There are 3 types of CRISPR systems which have been identified in both bacteria and Archaea [1], the most widely researched and characterised being the CRISPR associated nuclease protein of Streptococcus pyogenes which is called Cas9. The Cas9 protein from S. pyogenes contains 2 nuclease domains and is the most widely used Cas within gene editing research. After an initial viral infection, the cell will insert part of the viral DNA into the CRISPR loci of its own genetic material (where it is called a Spacer) and the fragment sits between palindromic repeats of non-coding DNA. In a recurrent infection, the CRISPR Array within the genome undergoes translation to generate crRNA and forms an effector complex containing the Cas protein and trans-activating CRISPR RNA (tracrRNA) [3]. When the effector complex interacts with the invading viral DNA, it binds to a Protospacer adjacent motif (PAM), the DNA unwinds and the crRNA binds to the complimentary sequence within the viral genome. The endonucleases within the Cas9 protein will then cleave the DNA, causing the neutralisation of the virus.
Interest in using the CRISPR Cas method for genome editing in research has grown exponentially in the last decade and the technique has a large scope as to what it can achieve. For example, the system can be used for the creation of knockout genes, gene silencing and for alterations in transcription and gene expression. Another use is to deactivate the nuclease enzymes and add a different enzyme so it can be transported to the target sequence. Furthermore, CRISPR can add a fluorescent tag to target sequences to see where a gene lies within a cell and aid in the visualisation of the 3D structure of the genome.
In recent publications, Douglas et al (2021) used a bicomponent CRISPR-Cas9 strategy to produce single sex mice litters. The team created single guide RNA (sgRNA) to target the Topoisomerase 1 gene as the absence of the gene would cause early embryonic lethality. They then created mouse lines expressing Cas9 on the X and Y chromosomes. When lines were mated, the resulting litters were male- or female-only with one hundred percent efficiency [4]. Thong et al (2021) used CRISPR Cas9 editing to rapidly produce new variants of the antibiotic Enduracidin. Gene editing was used to alter subdomains in nonribosomal peptide synthetase (NRPS) and was inserted into Streptomyces fungicidicus via plasmid. The results showed new variants of Enduracidin in sufficiently high yields and the process was found to be faster and more efficient than through conventional chemical processing [5]. Mertaoja et al (2021) studied sporulation in Clostridium botulinum, creating optimum anaerobic growth conditions using a Whitley Workstation. The research team used CRISPR Cas9, and the double stranded break repair mechanism called homology-directed repair (HDR), to replace targeted genes with a mutant “bookmark” sequence of their own design that could act as a sgRNA target for Cas9 [6]. The mutant strain could then be manipulated to study sporulation of the organism using different media types.
The CRISPR Cas system has the potential to revolutionize the treatment of genetic diseases and make improvements in farming and agriculture. One of the most promising areas of research is to increase the yields of crops by editing the genome and making crops more resistant to adverse conditions such as droughts and diseases.
Written by Charlotte Austin a microbiologist in our contract laboratory.
Charlotte Austin a microbiologist in our contract laboratory.
DNA/Genome Editing:
and CRISPR-Cas9?
Genome editing (also called gene editing) is a group of technologies that give scientists the ability to change an organism's DNA. These technologies allow genetic material to be added, removed, or altered at particular locations in the genome. Several approaches to genome editing have been developed. A well-known one is called CRISPR-Cas9, which is short for clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9. The CRISPR-Cas9 system has generated a lot of excitement in the scientific community because it is faster, cheaper, more accurate, and more efficient than other genome editing methods.
CRISPR-Cas9 was adapted from a naturally occurring genome editing system that bacteria use as an immune defense. When infected with viruses, bacteria capture small pieces of the viruses' DNA and insert them into their own DNA in a particular pattern to create segments known as CRISPR arrays. The CRISPR arrays allow the bacteria to "remember" the viruses (or closely related ones). If the viruses attack again, the bacteria produce RNA segments from the CRISPR arrays that recognize and attach to specific regions of the viruses' DNA. The bacteria then use Cas9 or a similar enzyme to cut the DNA apart, which disables the virus.
Researchers adapted this immune defense system to edit DNA. They create a small piece of RNA with a short "guide" sequence that attaches (binds) to a specific target sequence in a cell's DNA, much like the RNA segments bacteria produce from the CRISPR array. This guide RNA also attaches to the Cas9 enzyme. When introduced into cells, the guide RNA recognizes the intended DNA sequence, and the Cas9 enzyme cuts the DNA at the targeted location, mirroring the process in bacteria. Although Cas9 is the enzyme that is used most often, other enzymes (for example Cpf1) can also be used. Once the DNA is cut, researchers use the cell's own DNA repair machinery to add or delete pieces of genetic material, or to make changes to the DNA by replacing an existing segment with a customized DNA sequence.
Genome editing is of great interest in the prevention and treatment of human diseases. Currently, genome editing is used in cells and animal models in research labs to understand diseases. Scientists are still working to determine whether this approach is safe and effective for use in people. It is being explored in research and clinical trials for a wide variety of diseases, including single-gene disorders such as cystic fibrosis, hemophilia, and sickle cell disease. It also holds promise for the treatment and prevention of more complex diseases, such as cancer, heart disease, mental illness, and human immunodeficiency virus (HIV) infection.
Ethical concerns arise when genome editing, using technologies such as CRISPR-Cas9, is used to alter human genomes. Most of the changes introduced with genome editing are limited to somatic cells, which are cells other than egg and sperm cells (germline cells). These changes are isolated to only certain tissues and are not passed from one generation to the next. However, changes made to genes in egg or sperm cells or to the genes of an embryo could be passed to future generations. Germline cell and embryo genome editing bring up a number of ethical challenges, including whether it would be permissible to use this technology to enhance normal human traits (such as height or intelligence). Based on concerns about ethics and safety, germline cell and embryo genome editing are currently illegal in the United States and many other countries. https://medlineplus.gov/genetics.
Use of Genome editing: Basic research exploring human and nonhuman genomes is critical to help scientists understand the basic biology underlying disease, as well as to discover new possible therapeutic targets. Although excitement about the potential for gene therapy has grown tremendously since the discovery of CRISPR, the vast majority of work undertaken by scientists funded by NHGRI or other NIH Institutes takes place in the petri dish and in nonhuman organisms such as mice or zebrafish. Researchers rely on genome editing tools as a way to explore the connection between genotype (genes) and phenotype (traits). A typical study might be to model human disease in mice by deleting or editing certain genes that are thought to contribute to the disease. This approach can help researchers determine if specific changes made to the genome contribute to the disease. It can also lead to the creation of "disease models," or laboratory animals that mimic human diseases and can be studied to test new therapies.
In the clinic, there are proposals to use genome editing as a treatment for disease. Many diseases from cancer to asthma have genetic bases. Through the application of genome editing technologies, physicians might eventually be able to prescribe targeted gene therapy to make corrections to patient genomes and prevent, stop, or reverse disease.
Genome editing is currently being applied to research on cancer, mental health, rare diseases, and many other disease areas.
Basic research exploring human and nonhuman genomes is critical to help scientists understand the basic biology underlying disease, as well as to discover new possible therapeutic targets. Although excitement about the potential for gene therapy has grown tremendously since the discovery of CRISPR, the vast majority of work undertaken by scientists funded by NHGRI or other NIH Institutes takes place in the petri dish and in nonhuman organisms such as mice or zebrafish. Researchers rely on genome editing tools as a way to explore the connection between genotype (genes) and phenotype (traits). A typical study might be to model human disease in mice by deleting or editing certain genes that are thought to contribute to the disease. This approach can help researchers determine if specific changes made to the genome contribute to the disease. It can also lead to the creation of "disease models," or laboratory animals that mimic human diseases and can be studied to test new therapies.
In the clinic, there are proposals to use genome editing as a treatment for disease. Many diseases from cancer to asthma have genetic bases. Through the application of genome editing technologies, physicians might eventually be able to prescribe targeted gene therapy to make corrections to patient genomes and prevent, stop, or reverse disease. https://www.genome.gov/
Genome Profiling:
A laboratory method that uses a sample of tissue, blood, or other body fluid to learn about all the genes in a person or in a specific cell type, and the way those genes interact with each other and with the environment. Genomic profiling may be done to find out why some people get certain diseases while others do not. Genomic profiling may also be done on tumor tissue to look for mutations or other genetic changes in a tumor's DNA. This may help doctors understand how different types of cancer form and respond to treatment, which may lead to new ways to diagnose, treat, and prevent cancer. Also called genomic characterization. https://www.cancer.gov/publications.
NCI Dictionary terms from A to Z -Go to-https://www.cancer.gov/
Autosomal dominant inheritance is a way a genetic trait or condition can be passed down from parent to child. One copy of a mutated (changed) gene from one parent can cause the genetic condition. A child who has a parent with the mutated gene has a 50% chance of inheriting that mutated gene. Men and women are equally likely to have these mutations and sons and daughters are equally likely to inherit them.
https://www.cancer.gov/publications/dictionaries/genetics-dictionary
DNA fingerprinting is a laboratory technique used to determine the probable identity of a person based on the nucleotide sequences of certain regions of human DNA that are unique to individuals. DNA fingerprinting is used in a variety of situations, such as criminal investigations, other forensic purposes and paternity testing. In these situations, one aims to “match” two DNA fingerprints with one another, such as a DNA sample from a known person and one from an unknown person.
DNA fingerprinting. I think a lot of people are first introduced to DNA fingerprinting while watching crime shows. An officer collects some samples from the crime scene. They put it in a tube. And then an hour later, they hold up a brightly colored gel, squint at it, and say, aha, we have a match for the killer's DNA. Then the show is over. Of course, that isn't exactly how things work in real life. But DNA fingerprinting is an important part of forensic science. Although it can't really tell you exactly who committed a crime, it can be used to help narrow down a list of suspects based on how well their DNA matches the samples that were found at the crime scene. Investigators can also use the DNA results to search specific databases to find other potential suspects. By Lisa H.Chadwick (PhD).
Base Pairing
Deoxyribonucleic Acid (DNA)-DNA- PCR-
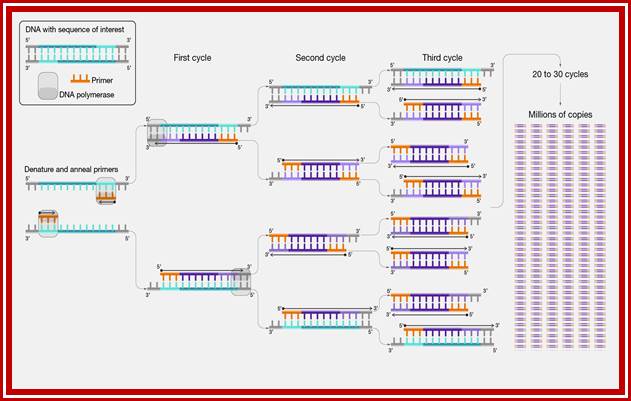
DNA- fragment subjected to Polymerase chain reaction (PCR) where, The DNA is replicated many times- 20 cycles and look at the number of copies produced (can be millions of copies, if you want).
Creating a DNA fingerprint involves a chemical test that analyzes the genetic makeup of a person or living thing and produces a pattern that is unique to that individual. It was created by Alec Jeffreys in the 1980s while he was at the University of Leicester in Leicester, UK. He discovered the technique after noticing that certain sequences of highly variable DNA, which do not contribute to genetic functions, are repeated within genes.
 An DNA fingerprinting
experiment. Look and Learn.
An DNA fingerprinting
experiment. Look and Learn.
After DNA is extracted from the sample, it is purified. Then, certain regions of the DNA are amplified using PCR. If longer segments of DNA are of interest, the amplified segments are usually separated in gel electrophoresis. Based on the size of DNA fragments, different DNA bands appear in a gel.
 PCR based DNA fingerprinting using gel
electrophoresis for separation. Wikipedia. CC-Attribution-Share Alike 4.0 International.
PCR based DNA fingerprinting using gel
electrophoresis for separation. Wikipedia. CC-Attribution-Share Alike 4.0 International.
For smaller segments, such as short tandem repeats (STRS), an STR analysis technique called capillary electrophoresis is used to separate and identify these very small fragments.
Process 3: Real-time PCR
Real-time PCR is accurate, cheap, reliable, and quick. It also goes one step further by quantifying the amount of DNA in a sample, making it ideal for crime scene investigations. It even works on very small amounts of DNA.
Fluorescence-labeled probes are used to bind to certain sections of DNA. The instrument reads the fluorescence output to determine how much that sequence is being amplified during PCR. Taller fluorescent peaks indicate that there are more repeats of that labeled sequence, which allows scientists to calculate the number of repeats in a sample.
DNA sequencing and capillary gel electrophoresis can be used in combination with this method.
The more these segments that are tested, the more accurate the DNA profile will be. The strips will show a barcode-like pattern that can then be compared to another sample of DNA to find a match.
DNA profiling is mostly used for legal and official purposes. This could include criminal investigations and court hearings. It is, however, not limited to these areas as profiling can be done for personal reasons. There have been increasing cases of parentage testing to confirm the paternal and child status between individuals.
Benefits:
This technique has benefits that range from solving crimes to finding family members and proving paternity.
Recently, genetic database sites have been implemented by law enforcement. One of the most famous examples is GedMatch, which was used to arrest the Golden State Killer in 2018. Joseph James DeAngelo is accused of killing 12 people and raping 45 women across California between 1976 and 1986. Police checked the crime scene DNA from this time against the genealogy sites and tracked him through a distant relative whose DNA partially matched evidence related to the serial killer, but with limitations.
PCR based DNA fingerprinting using gel electrophoresis for separation. Wikipedia. CC-Attribution-Share Alike 4.0 International.
Review of the Science of DNA Solves;
Purpose of DNA Fingerprinting
DNA profiling is mostly used for legal and official purposes. This could include criminal investigations and court hearings. It is, however, not limited to these areas as profiling can be done for personal reasons. There have been increasing cases of parentage testing to confirm the paternal and child status between individuals.
Benefits of DNA Fingerprinting;
This technique has benefits that range from solving crimes to finding family members and proving paternity. DNA’ solves uses the Othram technology to resolve cases and identify John Does and Mary Does. Othram is one of the most reliable and accurate test devices that the FBI and other investigative agencies use to fish out leads and resolve cases.
Who is who- descendants. / Criminals.
Nebula Genomics: This an unique bio-technology; It provides your ancestry, identify the criminals, and you can identify your genetic problems- of tissues and diseases.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Suspected Criminals PCR analysis.
Nebula Genomics:
At Nebula Genomics, we perform the most comprehensive 30X Whole Genome Sequencing to ensure that you can discover answers to all your genetic questions. This test sequences 100% of your DNA compared with just 0.2% offered by other companies.
Using the Nebula Research Library, you can discover research on the most up to date genetic discoveries and compare the results with your own genome. Through this service you can unlock more personal genetic information than ever before.
Nebula Genomics is the most affordable way to sequence your entire genome. Interested in learning more about your ancestry? We also offer deep ancestry insights through our partnership with Family Tree DNA: See what questions you can unlock by sequencing your DNA today! Check out our blog for more educational articles, including this one on the differences between DNA and RNA!.
Using PCR, portions of DNA that are known to contain VNTRs can be amplified. The resultant product is then viewed to obtain the individuals DNA genotype. In order to visualize the DNA profile from a given locus, the DNA fragments need to be separated according to their sizes using gel electrophoresis. An electric current is applied to an agarose gel matrix. Since DNA is negatively charged due to the phosphate groups, the fragments will move towards the positive pole. DNA molecules of different sizes migrate through the gel at different speeds, with the smaller fragments moving faster that the larger ones. Once the DNA is stained in the gel, it can be visualized to reveal the DNA profile.
Instruments used- one of them:
(Left) Fictitious agarose gel. Lanes 1, 2 and 3 represent bands using the data from the individuals listed above. Lane 4 represents DNA from a crime scene. (Right) Typical agarose gel apparatus, using Ethidium Bromide -provides color.
A Laser scan of the gel produces a genomic fingerprint of the type below:
Perhaps one of the most technological advanced techniques (according to my knowledge, I am 85 yrs. young)-.
https://www.chem.fsu.edu/chemlab/chm1020c/Lecture%2012/03.php